
El carcinoma de células de Merkel (CCM) fue descrito por primera vez por Toker 1 en 1972, con el nombre de “carcinoma trabecular de la piel”, proponiendo un origen sudoríparo o ecrino para este tumor, basado en su apariencia histológica. Seis años después, la demostración ultraestructural de gránulos neurosecretores llevó a Tang y a Toker a concluir que el carcinoma trabecular derivaba de la cresta neural y a sugerir la célula de Merkel como origen de esta lesión 2. Otros términos que se han propuesto y que reflejan el origen neuroendocrino de las células son “carcinoma de células de Merkel”, “carcinoma neuroendocrino primario de la piel”, “neoplasia neuroendocrina cutánea primaria” o “APUDoma cutáneo” 3,4. Los términos “carcinoma de células pequeñas de la piel” o “carcinoma extrapulmonar de la piel” reflejan su semejanza con el carcinoma de células pequeñas del pulmón. No obstante, la denominación más empleada en la literatura sigue siendo la de “carcinoma de células de Merkel”. El CCM es un tumor cutáneo poco habitual que suele presentarse en individuos de raza blanca y edad avanzada como una lesión nodular eritematosa de rápido crecimiento, localizada en la región de la cabeza y cuello, principalmente periorbitaria. Se asocia frecuentemente a otras neoplasias cutáneas. Su curso clínico es a menudo agresivo, con un elevado índice de recurrencias locales, diseminación linfática regional y metástasis a distancia 5, con una tasa de mortalidad directamente relacionada con el tumor de hasta un 65%. 6 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
EPIDEMIOLOGÍAEl CCM es un tumor poco frecuente (menos del 1% de todos los tumores cutáneos malignos, 100 veces menos frecuente que el melanoma) 7,8, con una incidencia total ajustada a la edad de 0.44 casos por cada 100,000 habitantes/año (año 2001) 9. En relación con la raza, la incidencia es de 0,23 por 100.000 para los pacientes de raza blanca y de 0,01 para los de raza negra, en EEUU (años 1986-1994) 10. La neoplasia se presenta en edades avanzadas, la mayoría de los casos entre las séptima y octava décadas de la vida (media: 69 años) y sólo el 5% antes de los 50 años 8. Sin embargo, la enfermedad ocurre a una edad inferior en sujetos inmunodeprimidos. Por ejemplo, en pacientes receptores de transplante, la edad media al diagnóstico es aproximadamente 10 años menor que la de los pacientes no inmunocomprometidos. En una serie, la edad media al diagnóstico fue de 53 años, y el 29% de los casos eran menores de 50 años de edad 11. En la mayoria de las series, la incidencia es algo mayor en varones, variando la proporción de 1,4-1.5:18,12 a 2-3:1 10. Algunos autores, sin embargo, han observado mayor incidencia en mujeres 13-17. El CCM se asocia con una elevada incidencia a otros tumores cutáneos y neoplasias hematológicas. En una publicación, 17 de 67 pacientes (25%) con CCM tenían una segunda neoplasia síncrona o metácrona, la mitad de las cuales eran carcinomas epidermoides 18. FACTORES ETIOPATOGÉNICOSLa etiología del CCM es desconocida, aunque la exposición crónica a la radiación ultravioleta y lainmunosupresión se proponen como principales factores etiopatogénicos implicados en el desarrollo de este carcinoma. En cuanto a la primera, hay una serie de evidencias que la apoyan: - La mayor incidencia en pacientes de raza blanca y en localizaciones fotoexpuestas 19. Autores como Miller et al 10 han encontrado relación entre el índice de radiación UVB y la incidencia de CCM. - La marcada asociación de los CCM con otros procesos cutáneos relacionados con el daño actínico, tales como queratosis actínica, carcinoma epidermoide, carcinoma basocelular o enfermedad de Bowen 20-22. - La aparición de CCM en pacientes en tratamiento con psoralenos y radiación ultravioleta A (PUVA) 23. - La publicación de una mutación en el gen p53 típicamente relacionada con la radiación ultravioleta B, similar a la descrita en el carcinoma basocelular y en el carcinoma epidermoide24. No obstante, dado que el CCM en ocasiones se presenta en áreas no fotoexpuestas 25, se postulan otros factores etiológicos entre los que destaca la inmunosupresión. Entre los hallazgos que avalan esta hipótesis se encuentran: - La demostración de casos de remisión espontánea, tanto del tumor como de las metástasis linfáticas regionales, presumiblemente inmunomediada. 25-35 - La asociación del CCM con situaciones de inmunodeficiencia, * Secundaria a la administración de tratamiento inmunosupresor en relación con otros procesos concomitantes: enfermedades autoinmunes 36,37, fracaso renal crónico 38, sarcoidosis pulmonar 39, trasplante renal y cardiaco 40-41. El riesgo de CCM en pacientes con transplante renal ha sido estimado en un 0.13/1000 habitantes año 42. Además, en pacientes post-trasplantados, la relación melanoma-CCM es de 6:1, mientras que en la población general es de 65:1 11. * En relación con otras enfermedades concomitantes de por sí inmunosupresoras: pacientes infectados con el VIH 43,44, neoplasias hematológicas: leucemia 19, mieloma múltiple 45, linfoma no Hodgkin 12,46. El riesgo relativo de desarrollar CCM en individuos con VIH ha sido calculado en 13,4/100,000 44. * Además, el CCM puede estar asociado con otros tumores no cutáneos en un 25% de los pacientes, como de mama, ovarios y cabeza y cuello 18, que pueden favorecer una inmunosupresión secundaria, bien por los fármacos inmunosupresores o por las neoplasias en sí mismas. La exposición a arsénico 47, y su coincidencia con displasia ectodérmica congénita y enfermedad de Cowden ha sido también señalados en diversas publicaciones 48. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
El CCM se presenta típicamente como una lesión cutánea primaria (75%) 8, aunque puede debutar como una enfermedad metastásica. La lesión primaria suele ser indolora, firme, sobreelevada, de crecimiento rápido, que aparece como un nódulo o pápula solitaria rojo-violácea con telangiectasias en superficie (Fig.1), por lo que en ocasiones se confunden con carcinomas basocelulares, carcinomas epidermoides, melanomas amelanóticos, hemangiomas y linfomas cutáneos 20,49-50. Con frecuencia, el CCM no se incluye en el diagnóstico clínico inicial. Su tamaño habitual oscila entre 0,5 y 5 cm de tamaño (generalmente, menor de 2 cm) y no suele estar ulcerado. Puede evolucionar a una placa rojo-violácea mal definida 51. Se han descrito algunas formas gigantes, de hasta 23 cm 52. Se localiza sobre todo en zonas expuestas al sol de la cabeza y cuello (47%) (Fig.2), siguiendo en orden de frecuencia las extremidades (33%) (Fig.3), el tronco (10%), los genitales y la región perianal (2%) y otros (menos del 1%) 53,55. Una distribución similar ha sido observada en un revisión de 1034 casos 8. En la cabeza y cuello, las mejillas y los párpados son las áreas más comúnmente afectadas 54. Se han descrito diversas formas clínicas de presentación atípica como son en forma de una mínima ulceración en la punta de la nariz 56, como nódulos subcutáneos en la región inguinal 57, tejido de granulación en tejido del pie de un adolescente 58, una lesión en el pezón de una mujer 59, o una placa eritematosa que semeja un angiosarcoma 39,60. La mayor parte de los pacientes debutan como enfermedad localizada al diagnóstico (76-89%), con afectación ganglionar entre el 10-18% y metástasis a distancia entre un 1-2% 53,61-62. Sin embargo, se trata de un tumor agresivo, con una tasa de recurrencia de entre el 33 y el 44%; con afectación ganglionar locorregional hasta en el 55-60% de los casos y metástasis a distancia hasta en el 33% (piel, hígado, pulmón, hueso y cerebro) 19-20-61,63. Como se ha señalado previamente, se encuentra descrita la regresión espontánea del tumor. Hasta el momento se han documentado 7 casos de regresión espontánea completa primaria (después de biopsia incisional) 25-30 y 5 casos más en los que la regresión sucedió después de recurrencia local o regional de la neoplasia (regresión espontánea completa secundaria) 31-35. Hasta un 10-20% de los casos se presentan sin tumor primario conocido 64-65, lo que parece encontrarse asociado a un mejor pronóstico 66. 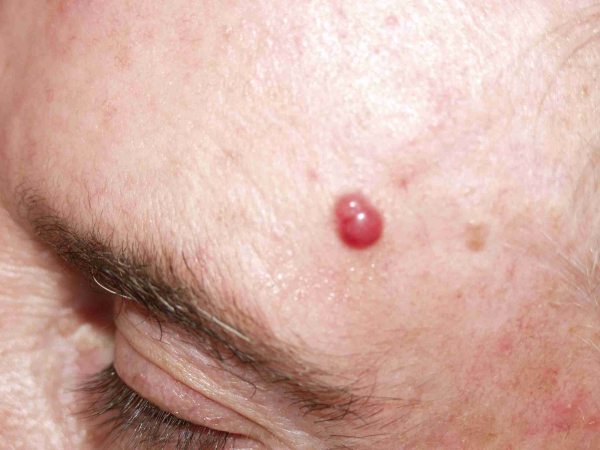 Figura 1: Pápula eritematosa en la frente (paciente 1) 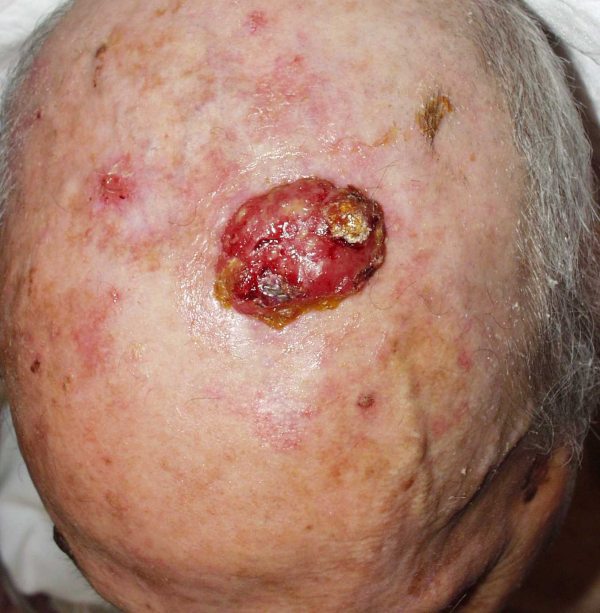 Figura 2: Tumor en el cuero cabelludo (paciente 2) 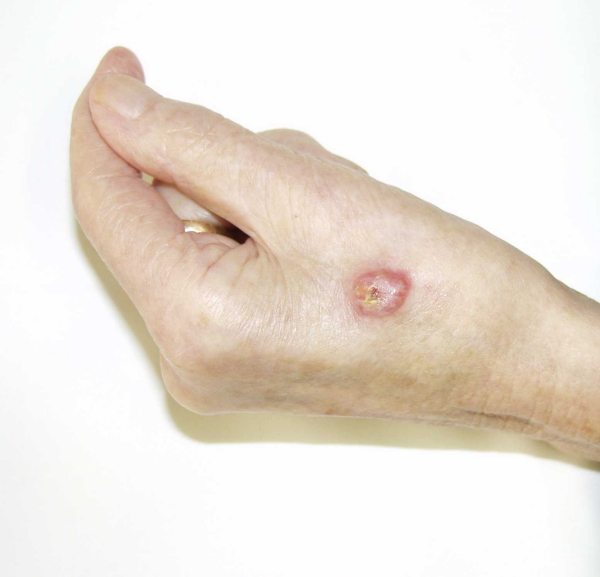 Figura 3: Nódulo en el dorso de la mano (paciente 3) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
El tumor se localiza en la dermis (Fig.4), y puede extenderse al tejido celular subcutáneo y a los tejidos blandos profundos. La epidermis suele encontrarse respetada (Fig.5), aunque puede estar ulcerada 67,68 (Fig.6), o mostrar hiperplasia reactiva. Se se han descrito casos de epidermotropismo, en algunas series hasta en un 30% de los casos 22, o de extensión “pagetoide” 69, e incluso se han publicado tres casos exclusivamente epidérmicos 70-71,54. En estos casos, el CCM puede simular un melanoma, una enfermedad de Paget extramamaria, una micosis fungoide, una enfermedad de Bowen pagetoide o un epitelioma intraepidérmico 69. Estos casos con epidermotropismo marcado se han empleado como evidencia de un origen en la célula de Merkel. Los bordes del tumor pueden ser infiltrativos o expansivos (Fig.4). Aunque es un hallazgo infrecuente, se han descrito tumores encapsulados. El CCM está constituído por células pequeñas y monomorfas (5-20µm de diámetro), de morfología redondeada u ovoidea y bordes mal definidos. Tienen escaso citoplasma basófilo y un núcleo vesicular e hipercromático con cromatina dispersa finamente granular y múltiples nucleolos de pequeño tamaño (Fig.7). De forma casi constante, suele existir un elevado índice mitósico (Fig.8) que oscila entre 3 y 15 mitosis por campo de gran aumento 1,45,68,72-74. Son frecuentes los fenómenos de apoptosis, pudiendo observarse necrosis focal en tumores grandes (Fig.9). La triada de núcleo vesicular con pequeño nucleolo, abundantes mitosis y apoptosis es característico del CCM 48,50. En ocasiones se observan focos de diferenciación escamosa (Fig.10) o ecrina. Esta tendencia, así como la asociación del CCM con otras neoplasias epiteliales como el carcinoma epidermoide, infiltrante o in situ, la queratosis actínica o el carcinoma basocelular, sugieren un nexo entre el CCM y el epitelio 22. Son extremadamente infrecuentes otros patrones de diferenciación como músculo esquelético, leiomiosarcoma, linfoepitelioma-like y fibroxantoma atípico-like. Se ha también descrito diferenciación rabdomioblástica en una metástasis ganglionar 75. Es frecuente observar un infiltrado inflamatorio linfoplasmocitario dentro y en la periferia del tumor, siendo rara la reacción desmoplásica del estroma 76. A menudo se observan imágenes de invasión vascular o linfática 72,77-78 (Fig.11). Se ha descrito la presencia de incrustación basofílica (depósito de DNA) alrededor de los vasos intratumorales (fenómeno de Azzopardi) 79. Con técnicas histoquímicas las células tumorales muestran reacción argirófila (Grimelius) positiva pero son argentafin 32, PAS y Sudán negro negativas. Los tumores que han regresado completamente se caracterizan por presentar un infiltrado inflamatorio linfocitario perivascular, ocasionales folículos linfoides, un número variable de histiocitos espumosos y fibrosis, sin celularidad tumoral residual 29. En los casos de regresión parcial se observa un infiltrado linfoide más denso 80. Tipos histológicos Gould estableció 3 patrones histológicos en el CCM 81: trabecular, de célula pequeña y de célula intermedia. - Patrón trabecular (Fig.12 y Fig.13): descrito por primera vez por Toker en 1972 1, es el patrón mejor diferenciado y el menos frecuente de los tres, observándose en menos de la cuarta parte de los casos publicados. Se caracteriza por células pequeñas, redondas o poligonales, que muestran un núcleo grande y ovalado con nucleolo prominente, dispuestas en un patrón organoide de trabéculas sólidas, irregulares y anastomosadas que pueden exhibir formaciones pseudoglandulares. En raras ocasiones se observan pseudorosetas, descritas por Toker en su artículo original. En esta variante, el índice mitósico es escaso o moderado. - Patrón de célula intermedia (Fig.14): es el más frecuente (50% de los casos). Se trata de un patrón difuso caracterizado por grandes nidos sólidos de células tumorales separados por finos tractos de tejido conectivo y focos de necrosis. Suelen presentar un número elevado de mitosis. Hay un infiltrado linfocitario dentro y alrededor del tumor. Los tumores que adoptan este patrón de crecimiento pueden situarse próximos a los anejos y conectar con la epidermis. - Patrón de célula pequeña (Fig.15 y Fig.16): Se caracteriza por sábanas y grandes grupos intradérmicos de células pequeñas e hipercromáticas, sin diferenciación glandular. A menudo muestran áreas de necrosis y se asemejan a otros tumores indiferenciados de célula pequeña. Con gran frecuencia se observan tumores conformados por células de diferentes tamaños adoptando diversos patrones de crecimiento, siendo imposible encasillarlos en un tipo histológico concreto. El CCM puede presentar gran similitud macroscópica e histológica con otras lesiones, incluyendo el carcinoma basocelular, el carcinoma epidermoide, el carcinoma metastásico de células pequeñas del pulmón u otro origen, algunos linfomas cutáneos, el neuroblastoma del adulto, el carcinoide metastásico, el melanoma amelanótico, neoplasias anexiales, el rabdomiosarcoma, el osteosarcoma de células pequeñas, la histiocitosis de células de Langerhans, el dermatofibrosarcoma protuberans y el sarcoma de Ewing 82,83. El estudio histopatológico e inmunohistoquímico y, en algunos casos, ultraestructural permiten realizar el diagnóstico diferencial Inmunohistoquímica El CCM expresa marcadores de origen epitelial y de tipo neuroendocrino. Entre los primeros destacan, fundamentalmente, las queratinas de bajo peso molecular (Fig.17) como CK8, CK18, CK19 y CK20. De todos ellos la más útil es la CK20 (Fig.18), positiva en el 97% de los casos 84,85. Aunque este marcador también es positivo en otros tumores de células pequeñas como los de origen pulmonar (0,03%), cervical (9%) y salivar (60%), en las células del CCM, y también en las los células de Merkel normales, es característico el patrón típico de tinción de citoqueratinas (y también de neurofilamentos) en forma de acumulos globulares en situación paranuclear 86,87. La CK20 también se usa en la detección de micrometástasis ganglionares, fundamentalmente en el ganglio centinela 88. En relación con el empleo de la CK20, ha adquirido una gran importancia el TTF-1 (thyroid transcription factor-1), factor de transcripción nuclear expresado por células del tiroides y del pulmón, y por la mayoría de los carcinomas epidermoides. Este marcador se expresa también en adenocarcinomas (72.5%), carcinomas de células pequeñas (83%-100%), tumores carcinoides atípicos (100%) y carcinomas neuroendocrinos (75%) 89. No es expresado, sin embargo, por los CCM. Los datos iniciales publicados por Byrd-Gloster et al 90 señalan que el TTF-1 es expresado por el 97% de los carcinomas de células pequeñas del pulmón, pero no por los CCM. En el mismo estudio, la CK 20 etiqueta el 76% de los CCM, mientras que es expresada únicamente por el 3% de los carcinomas de células pequeñas del pulmón. Otro estudio ha señalado la expresión de TTF-1 en el 82.7% de los carcinomas de células pequeñas del pulmón, en el 42% de los carcinomas de células pequeñas extrapulmonares y en el 0% de los CCM 91. De este modo, una combinación de TTF-1 y CK 20 debería proporcionar la mayor sensibilidad y especificidad para distinguir entre CCM y otros carcinomas de células pequeñas, aunque no diferencie, en estos últimos, entre un origen pulmonar o extrapulmonar 89. La CK7 también resulta típicamente negativa, por lo que, en combinación con CK20 y TTF-1 puede ayudar a diferenciar el CCM de metástasis cutáneas de un carcinoma bronquial de células pequeñas 91,92. Otros antígenos derivados del epitelio, como el EMA, son expresados hasta en un 90% de casos de CCM 48,67. Las desmoplaquinas se expresan hasta en el 50% de los casos de CCM, mientras que se aprecia ausencia de expresión de CEA. Las células tumorales también son positivas para Ber-EP4, un anticuerpo dirigido directamente contra un antígeno de membrana específico de epitelio, y contienen a menudo altos niveles de proteína bcl-2 93. Los marcadores de diferenciación neuroendocrina también pueden emplearse para caracterizar este tumor. El más constante (50%-100%) de todos ellos es la NSE (Fig.19), que reacciona con intensidad variable 48,94. Las cromograninas B y A son positivas en el 100% y el 72% de los tumores respectivamente 95. La secretoneunina, derivada de la secretogranina II, se expresa en el 22% de los casos y la sinaptofisina (Fig.20) en el 39%. También se ha visto positividad para otros neuropéptidos como son la ACTH, sustancia P, gastrina, péptido intestinal vasoactivo, somatostatina y calcitonina 87. Las células del CCM también tienen la propiedad de expresar marcadores neurales como neurofilamento L y polipéptido M. La coexpresión de citoqueratinas y neurofilamentos se ha observado hasta en un 100% de casos de CCM 48. Los neurofilamentos son positivos también en los neuroblastomas, feocromocitomas, meduloblastomas y ganglioneuromas, pero estos tumores no muestran expresión para citoqueratinas. Pueden resultar útiles también para diferenciarlos de otros carcinomas neuroendocrinos y carcinoides, que muestran positividad para citoqueratinas pero no expresan neurofilamentos. No se incluyen los carcinomas de células pequeñas del pulmón, los carcinoides bronquiales y los tumores de células de los islotes del páncreas, que muestran coexpresión de ambos marcadores. La positividad para CD99 sugiere un PNET; sin embargo, en una serie publicada por Nicholson et al 97, un 40% de los CCM eran positivos para CD99, aunque a nivel citoplásmico. La detección inmunohistoquímica de CD117 se aprecia en un 95% de los CCM, pero no se corresponde con un curso más agresivo ni supone mutaciones en el receptor c-kit, por lo que no debería inducir a ensayos terapéuticos con inhibidores de la tirosin quinasa. Permite, sin embargo, diferenciar el CCM de una leucemia mieloide cutis 98. Algunos autores han señalado que la inmunoexpresión de CD44 puede indicar un potencial metastático 99. No se detectan proteína S-100, HMB-45 y NK 1/C3, útiles para el diagnóstico del melanoma. Tampoco se aprecia expresión para integrina, proteína ácida fibrilar glial, antígeno leucocitario común, actina, vimentina, laminina o metencefalina. Esta última es un marcador de las células de Merkel normales. En resumen, la detección inmunohistoquímica de filamentos intermedios, fundamentalmente CK20, TTF-1 (thyroid transcription factor-1) y marcadores neuroendocrinos en un tumor dérmico indiferenciado es útil en diferenciar CCM del carcinoma de células pequeñas metastásico y de otras neoplasias de morfología similar 20.92,100. El diagnóstico definitivo requiere negatividad para S-100, antígeno común leucocitario y citoqueratinas de alto peso molecular 85.94.
Aunque algunos autores opinan que con la clínica, la histología y la realización de estudios inmunohistoquímicos es suficiente para realizar un diagnóstico correcto de CCM 101, otros han sugerido que el diagnóstico debe confirmarse mediante microscopía electrónica siempre y cuando sea posible 1. Ultraestructuralmente, el CCM muestra agregados laxos de células células redondas y monomorfas embebidas en un estroma constituído por material finamente granular y fibras de colágeno dispersas. El núcleo es generalmente redondo u ovalado y, con la excepción de alguna pequeña indentación citoplasmática, presenta unos bordes lisos. Contiene una cromatina dispersa (eucromatina) y uno o varios nucleolos redondos de pequeño tamaño, a veces vacuolados, en localización excéntrica y con frecuencia adosados a la membrana nuclear. El núcleo se encuentra rodeado de una escasa cantidad de citoplasma excepto en el polo de la célula ocupado por un aparato de Golgi moderadamente desarrollado. El citoplasma contiene mitocondrias dispersas y cisternas de retículo endoplásmico rugoso. Además de las organelas habituales, también se observan gránulos electrodensos neurosecretores de 20-200 nm limitados por una membrana, localizados en la región del Golgi y en el ectoplasma. La presencia de estos gránulos es indispensable para el diagnóstico aunque hay que tener en cuenta que también pueden observarse en los carcinomas metastáticos de células pequeñas y que tienden a perderse en el material fijado en parafina. Otro hallazgo característico es la presencia deagregados paranucleares de filamentos intermedios de 10 nm. Y prolongaciones citoplásmicas cortas. También pueden observarse mecanoreceptores, uniones intercelulares complejas similares a desmosomas primitivos o uniones intermedias, y tonofilamentos. La identificación citológica de un carcinoma de células de Merkel requiere cierta experiencia, una adecuada correlación clínicopatológica y, frecuentemente, el empleo de técnicas de inmunocitoquímica o microscopía elecrónica. En general, los extendidos de CCM muestran abundante celularidad (Fig.21) que se dispone predominantemente de forma aislada o en pequeños grupos discohesivos que pueden mostrar moldeamiento nuclear (Fig. 22) y raras veces formar pseudorrosetas. Las células tumorales son de tamaño pequeño-intermedio, y presentan un núcleo monomorfo, redondeado u ovalado, con cromatina finamente granular de aspecto pulverulento y múltiples (2-5) nucleolos de pequeño tamaño (Fig.23). El citoplasma es escaso, generalmente en forma de un delgado ribete. Se observan frecuentes figuras de mitosis (Fig.24), algunas atípicas, y apoptosis. Ocasionalmente pueden existir células con un mayor pleomorfismo que oscila entre células con hendiduras, indentaciones y protrusiones de la membrana nuclear hasta células de aspecto abigarrado o multinucleadas. Domagala et al 117 describieron la presencia de “gotas” citoplasmáticas perinucleares, de coloración rosa pálido, homogéneas, relativamente densas, visibles al microscopio de luz en material de punción fijado en alcohol y teñido con H y E, que resultan positivas para filamentos intermedios de citoqueratina. Este hallazgo no ha sido descrito por todos los autores habiéndose sugerido que pudiera ser artefactual y relacionado con la tinción 118. En los casos en que existe un componente escamoso en el tumor, a veces es posible apreciarlo en el material aspirado (Fig.25), lo que dificulta el diagnóstico diferencial con el carcinoma epidermoide, fundamentalmente en aquellos pacientes que debutan con metástasis ganglionares sin tumor primario conocido. Tal y como se ha señalado previamente, la inmunocitoquímica ayuda a confirmar el diagnóstico y a descartar otros. Desde un punto de vista citológico, el diagnóstico diferencial de un aspirado de CCM se centra, fundamentalmente, en las neoplasias con patrón monomorfo de células pequeñas: La distinción con un carcinoma indiferenciado de células pequeñas de origen pulmonar puede ser imposible 119. Estos suelen presentar mayor fragilidad celular con imágenes de estiramiento cromatínico y moldeamiento nuclear. La presentación en forma de células aisladas, con ausencia de grupos celulares cohesivos y de cuerpos linfoglandulares en el fondo permiten la distinción con el linfoma no Hodgkin 119,116. La PAAF de carcinoma basocelular habitualmente muestra grupos fuertemente cohesivos de células con empalizada nuclear, ausentes en el CCM 116. A diferencia del CCM, el rabdomiosarcoma se caracteriza por extendidos con frecuentes células bi o multinucleadas y pequeños agregados ocasionales de células con fragmentos de tejido conectivo 116. Las extensiones citológicas de melanoma, pueden mostrar patrones muy variados, incluido el indiferenciado de célula pequeña. Deben buscarse células con citoplasma más abundante, con ocasional pigmento melánico y macronucleolo 111,116. Al igual que el CCM, el tumor neuroectodérmico primitivo se caracteriza por células pequeñas, monomorfas y discohesivas con cromatina “en sal y pimienta”. No obstante, en el tumor neuroectodérmico primitivo las células tumorales pueden formar rosetas, rara vez presentes en el CCM 116. ALTERACIONES GENÉTICASEl cambio citogenético más frecuente es la pérdida de heterozigosidad debido a traslocaciones y delecciones en el brazo corto del cromosoma 1 (1p36) (40% de los casos) 120,121. Los estudios de delección apuntan a más de un posible locus supresor en la región 1p32–1p36, aunque aún no se han identificado los genes candidatos 122. Se aprecian anomalías semejantes en tumores originados en la cresta neural como melanoma, neuroblastoma y feocromocitoma 120,123, lo que ha sido empleado como argumento a favor del origen neurocrístico del CCM 124. Se han observado similitudes entre el CCM y el carcinoma de células pequeñas del pulmón por la pérdida de heterozigosidad en 3p21, una región que se ve comúnmente afectada en el carcinoma de células pequeñas 123. Otras alteraciones cromosómicas frecuentes son la delección de 3p (46%), 5q (21%), 8p (21%), 10q (33%), 11q (17%), 13q (33%) y 17p (25%), y la ganancia de 1q (63%), 3q (33%), 5p (38%), 8q (38%), 19 (63%) y X (41%) 125,121. Ganancias menos frecuentes afectan a los cromosomas 6, 7, 20 y 21. En este tumor las amplificaciones son raras y, aunque no han podido demostrarse de forma convincente mutaciones específicas, se han descrito aberraciones en oncogenes y genes supresores como el bcl-2, p53 y PTEN 24,126,127. El bcl-2 es un oncogen antiapoptótico que se expresa en el CCM. Genes supresores como el p53 controlan el ciclo celular en células con daños en el DNA causados por la radiación ultravioleta. En un 20% de los casos de CCM se han detectado mutaciones en p53 que apuntan a que las mutaciones en este gen influyen en los estadios iniciales o en la progresión de algunos CCM. El gen p73 es otro gen estructural y funcionalmente similar al p53 ubicado en el cromosoma 1p36.33 (una región frecuentemente deleccionada en el CCM) que raras veces se muta en el CCM. También se han descrito mutaciones del gen supresor PTEN. A pesar de su marcada diferenciación neuroendocrina, el CCM comparte con otros tipos de carcinoma actividad de telomerasa, inexistente en células somáticas diferenciadas 128. Por otra parte, el CCM expresa varias moléculas de adhesión que podrían servir en un futuro como marcadores diagnósticos de este tumor como CD171 (L1CAM), CD24 y CD56, o moléculas de adhesión neuronal (NCAM) 129,130. En conclusión, las alteraciones genéticas asociadas con el desarrollo y progresión del CCM no son del todo conocidas. Existen anomalías citogenéticas entre el 30% y el 47 % de los casos; la más frecuente es la pérdida de heterozigosidad debida a translocaciones y deleciones en el cromosoma 1. Hasta la fecha, no ha sido implicado de forma concluyente ningún oncogen o gen supresor 50. 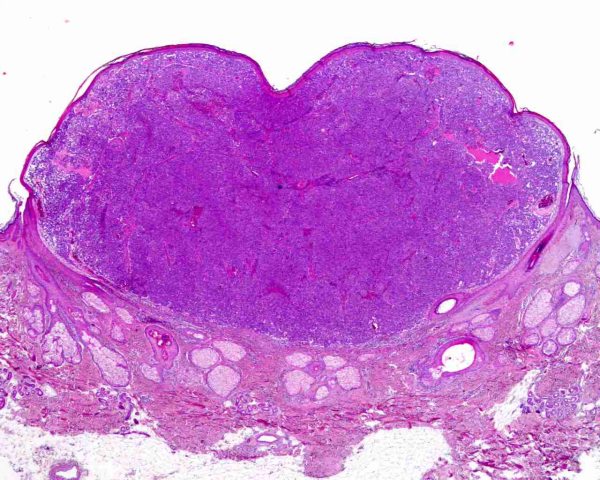 Figura 4: HE 20x. Tumor “azul”, localizado en la dermis. 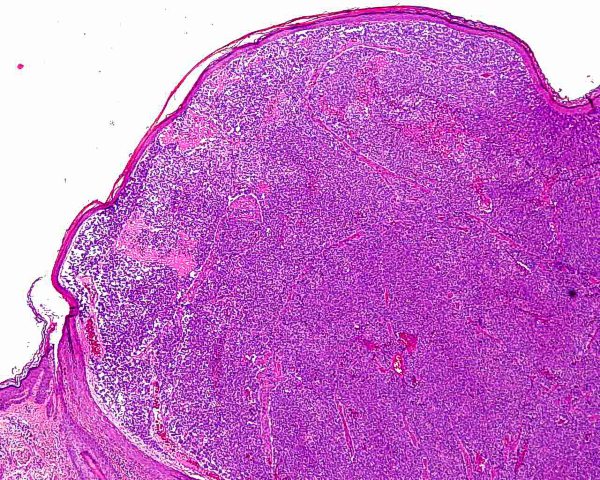 Figura 5: HE 40x. La neoplasia respeta, generalmente, la epidermis 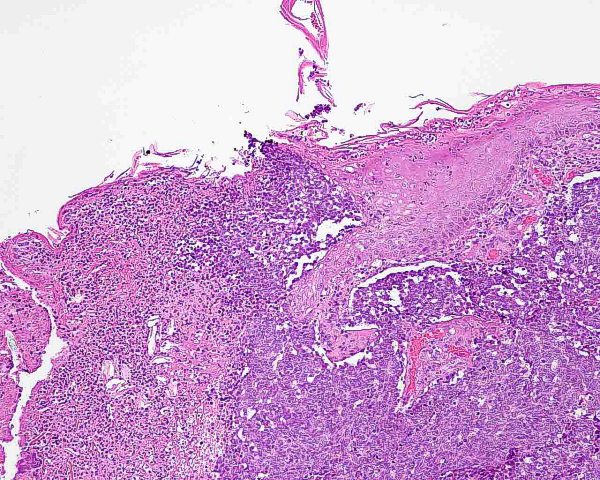 Figura 6: HE 100x. La epidermis puede estar ulcerada. 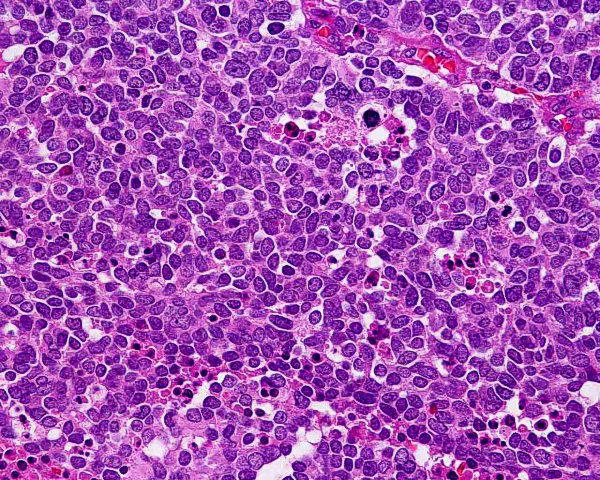 Figura 7: HE 400x. Las células son pequeñas y monomorfas, con núcleo vesicular e hipercromático con cromatina finamente granular y escaso citoplasma. 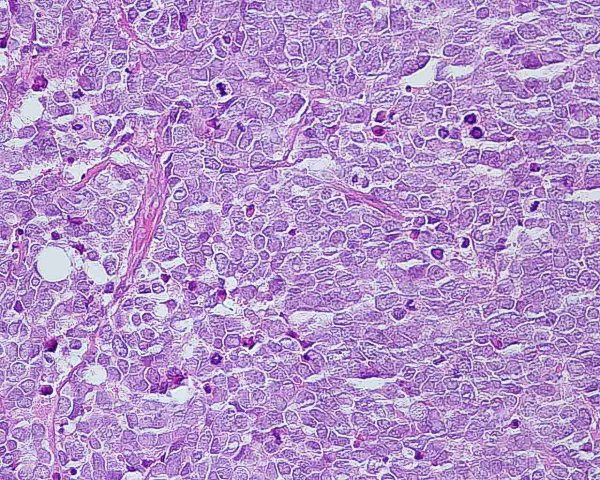 Figura 8: HE 400x. El tumor muestra un elevado índice mitósico 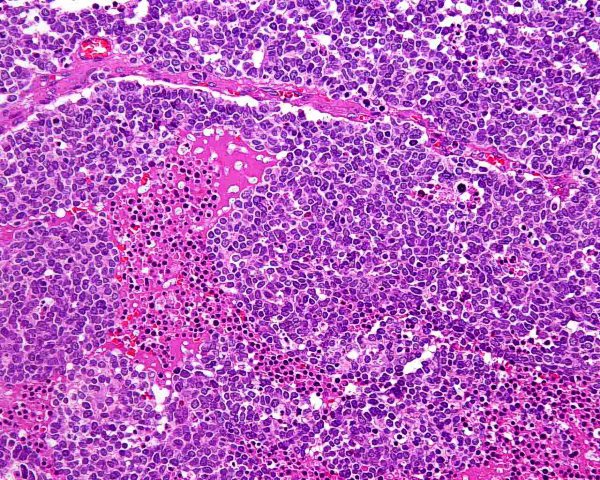 Figura 9: HE 200x. La apoptosis y la necrosis focal son fenómenos frecuentes. 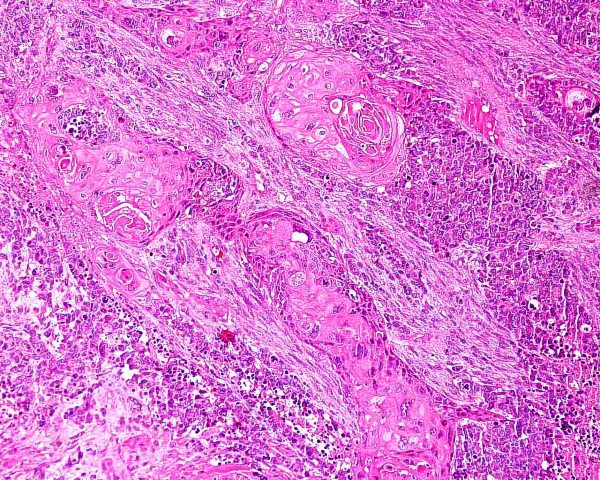 Figura 10: HE 100x. Componente escamoso del CCM. .jpg) Figura 11: HE 400x. La invasión linfovascular es un hallazgo casi constante. 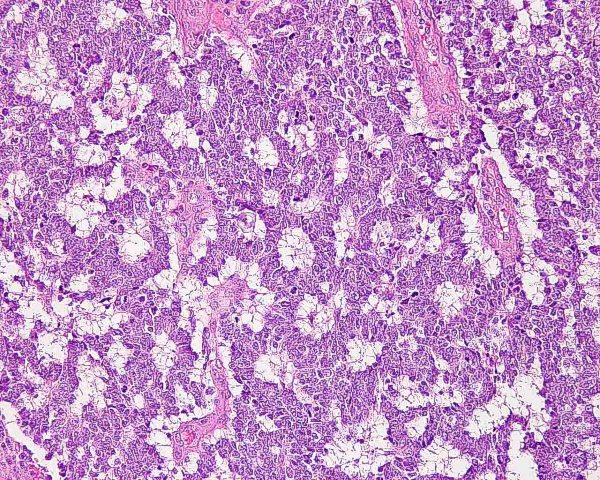 Figura 12: HE 200x. Patrón trabecular: Patrón organoide de trabéculas anastomosadas. 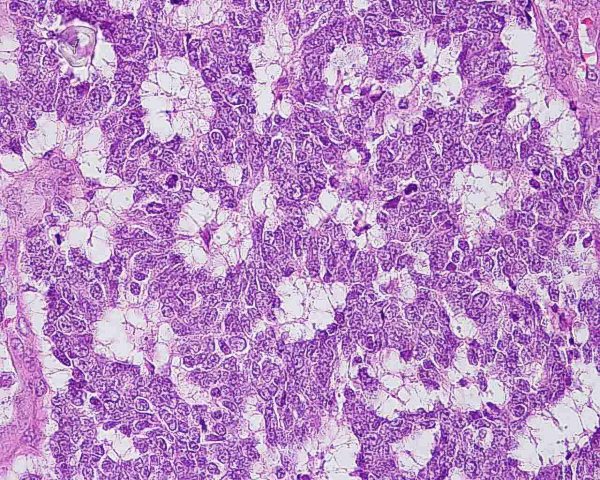 Figura 13: HE 400x. Patrón trabecular. Detalle de la anterior. 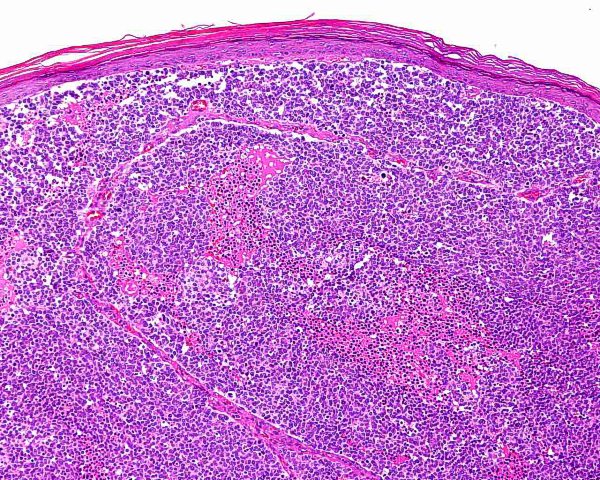 Figura 14: HE 100x. Patrón de célula intermedia. Patrón difuso con grandes nidos sólidos separados por tractos de tejido conectivo y focos de necrosis. 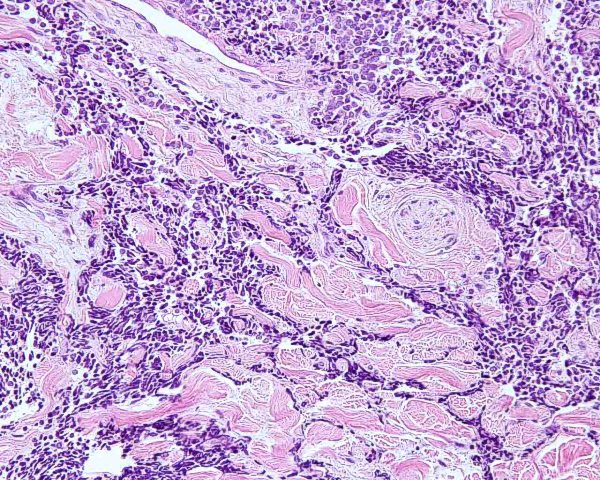 Figura 15: HE 200x. Patrón de célula pequeña. Grupos intradérmicos de células pequeñas e hipercromáticas, similares al carcinoma de células pequeñas de pulmón. 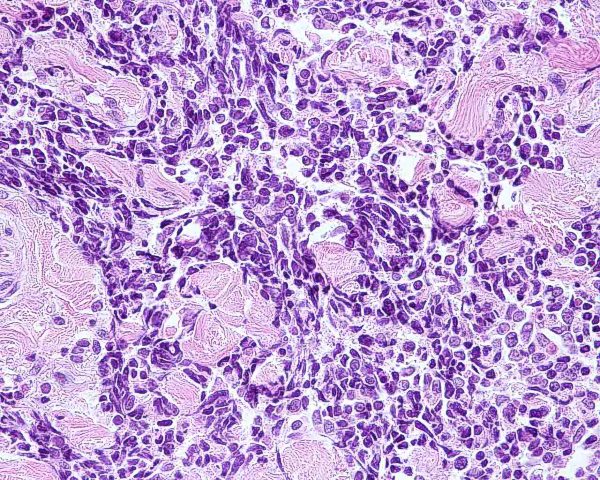 Figura 16: HE 400x. Patrón de célula pequeña. Detalle de la anterior. ![Figura 17: Cam 5.2 (CK 8, [18]) 200x. Marcaje en gota paranuclear.](http://www.conganat.org/7congreso/imagenes_trabajos/526-fig17.jpg) Figura 17: Cam 5.2 (CK 8, [18]) 200x. Marcaje en gota paranuclear. 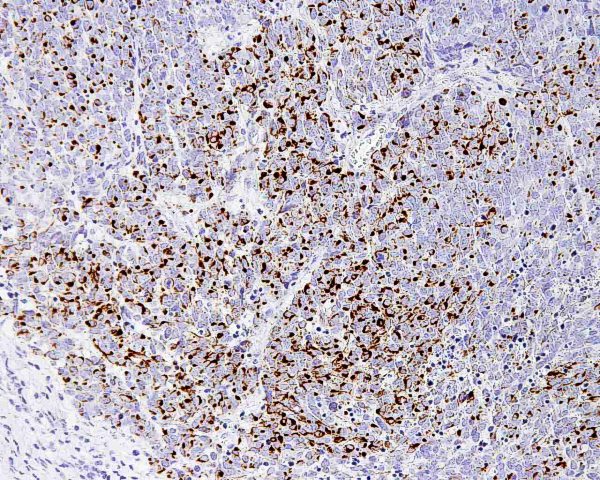 Figura 18: CK20 200x. Positiva en el 97% de los casos. Marcaje en gota paranuclear. 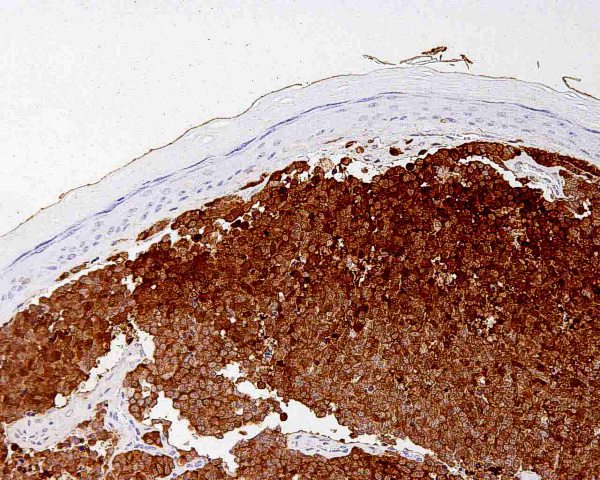 Figura 19: NSE 200x. Positiva en el 50-100% de los casos. 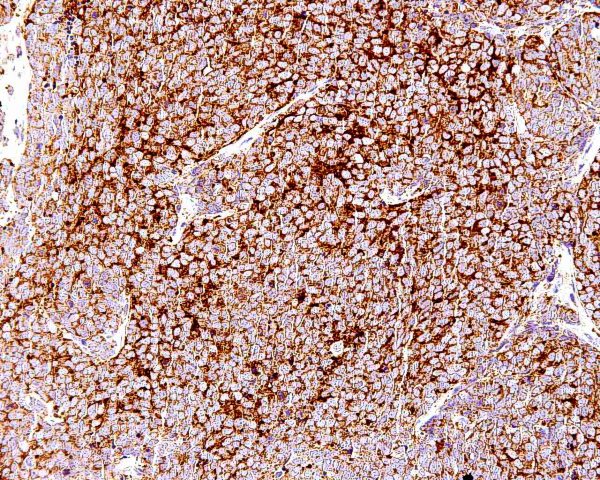 Figura 20: Sinaptofisina 200x. Positiva en el 39% de los casos. 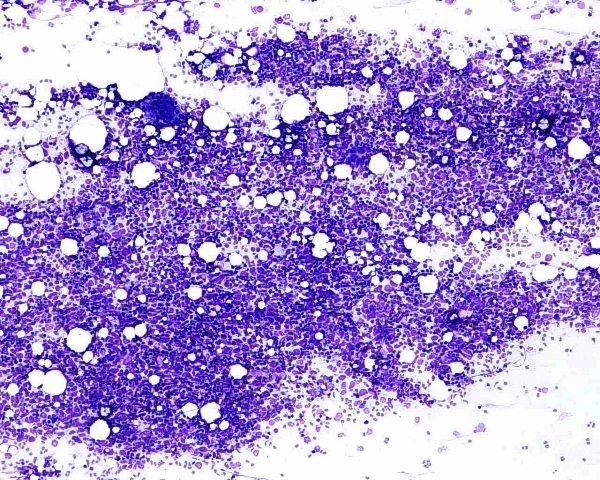 Figura 21: Diff Quick 100x. Los extendidos muestran abundante celularidad. 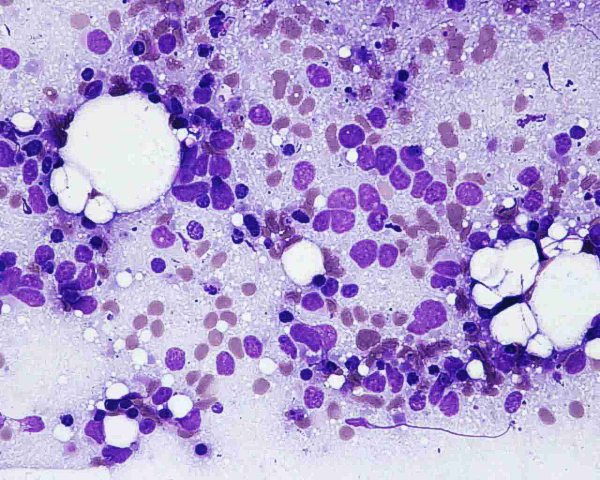 Figura 22: Diff Quick 400x. Las células se disponen de forma aislada o en pequeños grupos discohesivos que pueden mostrar moldeamiento nuclear. 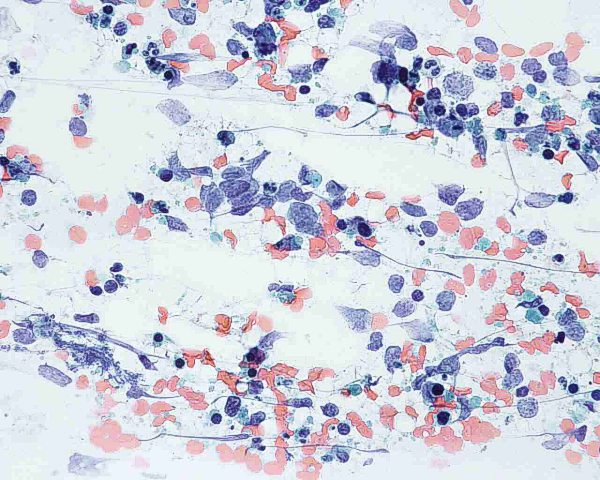 Figura 23: Papanicolaou 400x. Las células son de tamaño pequeño-intermedio con núcleo con cromatina finamente granular. Es habitual la apoptosis. 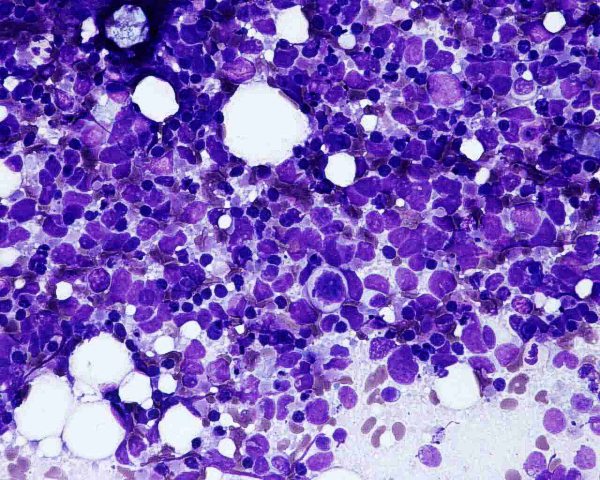 Figura 24: Diff Quick 400x. Son frecuentes las mitosis. 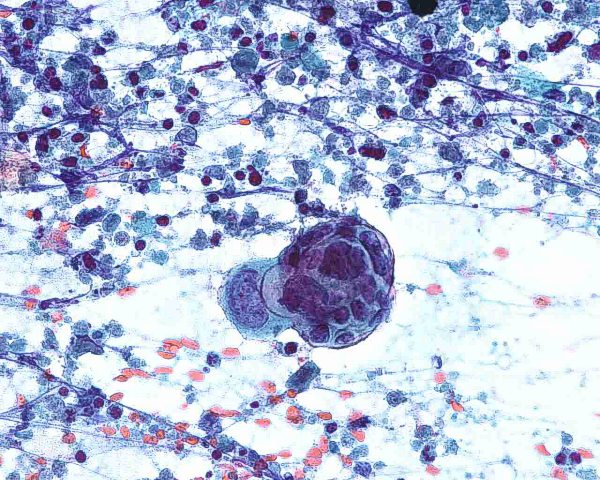 Figura 25: Papanicolaou 400x. A veces se aspira también el componente escamoso, lo que dificulta el diagnóstico diferencial con el carcinoma epidermoide. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Las células de Merkel fueron descritas por primera vez por Merkel en 1875 131 y se ubican en la capa basal de la epidermis y de los folículos pilosos. Su densidad ha sido estimada en 5-100 células/mm2 en la capa basal 132. Aunque pueden ser encontradas de forma aislada, tienden a aparecer en grupos, e íntimamente asociadas a fibras nerviosas mielinizadas existentes en las papilas dérmicas 133. El complejo célula de Merkel-axón es considerado actualmente el mecanorreceptor principal de la piel, motivo por el que estas células son más abundantes en los pulpejos de los dedos y la punta de la nariz. También son encontradas con mayor densidad en los labios y en la mucosa oral. La ultraestructura de la célula de Merkel es característica, con filamentos intermedios en el citoplasma, a veces en localización perinuclear; y gránulos densos rodeados por membrana, de 60-220 nm de diámetro, que recuerdan a los gránulos neurosecretores encontrados en las células APUD 20,73. El perfil inmunohistoquímico de las células de Merkel no tumorales es neuroendocrino (NSE, sinaptofisina y cromogranina) y epitelial (citoqueraqtinas de bajo peso molecular 8, 18, 19 y 20). El origen de la célula de Merkel es controvertido, aunque en los mamíferos existe la evidencia de que se originan a partir de la cresta neural 134, desde la que migran a la piel, donde se diferencian en células de Merkel maduras y expresan marcadores neurales y epiteliales. Origen del CCMLa histogénesis del CMM es un tema aún debatido. Entre las posibles células de origen se citan: - La célula de Merkel epidérmica, por sus similitudes ultraestructurales e inmunohistoquímicas - Un equivalente dérmico de la célula de Merkel, ya que el CCM se localiza habitualmente a este nivel. - Una célula del sistema APUD procedente de la cresta neural - Una célula madre epidérmica o anexial. La hipótesis del origen del CCM a partir de la célula de Merkel se apoya en argumentos como la positividad de ambos para NSE 135 y características ultraestructurales similares como son los gránulos neurosecretores, un núcleo lobulado con numerosos nucleolos y agregados paranucleares de filamentos intermedios 2,68,77,136. En contra de esta teoría está el hecho de que únicamente el 10% de los CMM se originan en la epidermis, siendo la gran mayoría intradérmicos, por lo que se ha postulado su origen en un equivalente dérmico de la célula de Merkel. Otra hipótesis sugiere un origen epitelial para el CCM basado en la positividad de las células tumorales para CEA (un marcador que solamente es positivo en los ductos ecrinos y en el acrosiringio) 70, Ber-EP4 (un anticuerpo monoclonal dirigido contra el antígeno epitelial de membrana) 137 y queratinas de alto y bajo peso molecular 138. También sustenta esta hipótesis la frecuente asociación del CCM con otros tumores epiteliales como el carcinoma epidermoide o la enfermedad de Bowen 137. Por último, algunos autores sostienen que el tumor se desarrolla a partir de células pluripotenciales con capacidad para diferenciarse tanto a células neuroendocrinas como epiteliales 72,137-139. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
ESTADIAJEAunque no existe un sistema de estadiaje universalmente aceptado, la mayor parte de los pacientes se estadian de acuerdo con el sistema introducido por el Memorial Sloan-Kettering Cancer Center 168 en los años 80: Estadio Ia: Confinado a la piel, <2 cm. de diámetro máximo Estadio Ib: Confinado a la piel, >=2 cm. de diámetro máximo Estadio II: Diseminación ganglionar regional Estadio III: Diseminación ganglionar más allá de los ganglios regionales y/o metástasis a distancia. Con arreglo a esta estadificación, la supervivencia a los 2 años en estudios recientes 53,140 es como sigue: Estadio Ia: 67% Estadio Ib: 59% Estadio II: 49% Estadio III: 23% En la actualidad, el sistema presenta 4 estadios para adaptarse a los sistemas habituales de estadificación del AJCC, y porque permite establecer dos grupos de riesgo (bajo –I y II- y alto –III y IV) 141: Estadio I: Confinado a la piel, tumor <2 cm. de diámetro máximo Estadio II: Confinado a la piel, tumor >=2 cm. de diámetro máximo Estadio III: Diseminación ganglionar Estadio IV: Metástasis a distancia. Para el estadiaje deben realizarse RS de tórax y TAC toracoabdominal. El empleo rutinario de TAC craneal es controvertido en pacientes asintomáticos. Las pruebas de imagen a nivel torácico se emplean, fundamentalmente, para poder diferenciar la lesión de la metástasis cutánea de un carcinoma de células pequeñas de pulmón. Algunos autores 141 recomiendan la estadificación ganglionar mediante biopsia del ganglio centinela en los casos de enfermedad localizada al inicio. El empleo de este procedimiento permite una clasificación más exacta de los pacientes, por lo que se obtienen unas cifras de supervivencia a los 5 años mejoradas en relación con estudios previos: Estadio I: 81% Estadio II: 67% Estadio III: 52% Estadio IV: 11% TRATAMIENTONo existe consenso acerca de la terapéutica más adecuada para el CCM en los estadios iniciales, en particular sobre el tratamiento adyuvante postquirúrgico. El uso de quimioterapia en recurrencias y enfermedad metastásica se complica por la elevada edad de los pacientes, que tienden a tolerar peor el tratamiento 53. No obstante, puede resumirse, básicamente, en la siguiente: - Resección quirúrgica: Amplia excisión quirúrgica del tumor primario (márgenes sanos de 2-3 cms. en torno al tumor), que alcance en profundidad la fascia (Fig. 26). La excisión quirúrgica puede ser el único tratamiento 14,142,168 o combinarse con radioterapia adyuvante del lecho quirúrgico y ganglios de drenaje 101,143,143,168. Si el tumor se localiza en el pabellón auricular, algunos autores recomiendan realizar una parotidectomía superficial 15. - Cirugía de Mohs: La cirugía micrográfica de Mohs ha demostrado ventajas frente a la extirpación amplia en algunas series 145. Puede mejorar el control local del tumor debido a la evaluación histológica del 100% de los márgenes quirúrgicos, incluyendo el margen profundo. Este último es importante por la tendencia de estas neoplasias a mostrar un extenso crecimiento vertical, por lo que el margen profundo puede encontrarse afecto después de la cirugía. El empleo de radioterapia después de cirugía de Mohs puede contribuir a reducir el riesgo de recidiva locorregional, sobre todo en tumores grandes o recurrentes 69,145,146. - Linfadenectomía regional: La linfadenectomía regional terapéutica está indicada en aquellos pacientes con afectación ganglionar clínica o radiológica. Resulta controvertido, sin embargo, el empleo de radioterapia o disección ganglionar profiláctica, aunque, ya que la afectación locorregional después de la resección quirúrgica aislada se aproxima al 50% 168, algunos autores consideran que todos los pacientes deberían considerarse de alto riesgo e indicar dicha linfadenectomía 147. Algunos autores recomiendan el empleo de linfadenectomía profiláctica en aquellos casos localizados en cabeza y cuello 168, con diámetro tumoral >1.5 cm 19, o evidencia histológica de invasión vascular o linfática 61, aunque no existen datos fehacientes acerca de la mejoría de los pacientes incluidos en estos subgrupos con esta terapéutica. Actualmente, se aconseja realizar linfadenectomía regional sólo si se detectan adenopatías palpables 148 y si éstas no se detectan clinicamente realizar biopsia del ganglio centinela 51,141, ya que, al igual que en el melanoma, la práctica de la linfadenectomía profiláctica no ha demostrado un incremento de la supervivencia 12 y sí de la morbilidad 14. - Ganglio centinela: Los datos relativos a la utilidad de esta técnica en el CCM son aún limitados 82,149,150. En una serie de 28 pacientes en los que se aplicó esta técnica, los 7 con ganglio centinela negativo en los que se realizó disección ganglionar adicional no mostraron depósitos tumorales en dichos ganglios 82. En 71 de los 177 pacientes (40%) de la serie de Allen et al 141 que debutaron con ganglios clínicamente negativos se realizó estadificación anatomopatológica, encontrándose afectación ganglionar en 16 (25%). Ya que no existen factores pronósticos que permitan predecir la diseminación ganglionar, esta técnica podría identificar a aquellos pacientes que se beneficiarían de la disección ganglionar regional. - Radioterapia: El CCM es un tumor radiosensible. El uso de la radioterapia incluye: · Tratamiento postoperatorio (adyuvante), del lecho quirúrgico y de los ganglios linfáticos regionales 151, con dosis entre 41-50 Gy, en 20-25 sesiones durante unas 5 semanas 140. Se ha descrito una disminución del riesgo local de recidivas con el uso de radioterapia adyuvante tras el tratamiento quirúrgico (10,5 % frente a 52,6%) y un aumento de la supervivenvia libre de enfermedad (88 frente a 58 meses) 152. Sin embargo, esta terapia combinada no ha demostrado un aumento significativo de la supervivencia global 16. · Tratamiento primario para tumores irresecables o pacientes inoperables, como tratamiento paliativo 153. · Radioterapia de rescate para enfermedad recurrente - Quimioterapia: El empleo de la quimioterapia puede ser considerado en las siguientes circunstancias: · Tratamiento postoperatorio (adyuvante): la quimioterapia adyuvante no parece disminuir los índices de recurrencia ni mejorar la supervivencia 21. · Tratamiento de la enfermedad de alto riesgo, asociada a radioterapia (Recurrencia; afectación ganglionar; tamaño tumoral >1 cm.; enfermedad residual macroscópica después de la cirugía) 66. · Tratamiento de las metástasis a distancia: Los agentes quimioterápicos habituales usados para CCM en estadios avanzados son los mismos que los usados para el carcinoma de células pequeñas de pulmón 53. La asociación con radioterapia parece que ofrece resultados más favorables 155. .jpg) Figura 26: Tras la biopsia, se realiza una exéresis amplia, con márgenes sanos de 2-3 cm. (paciente 1) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
PRONÓSTICOSe considera que el CCM tiene un curso similar a un melanoma de espesor intermedio o grueso, pero con un peor pronóstico 14. En la mayoría de los casos, el CCM es un tumor agresivo, con tendencia a la recidiva locorregional y a las metástasis ganglionares tempranas 149. Las recidivas pueden ser locales, regionales y a distancia: - Afectación ganglionar (33%) (Fig.30), a los 7-8 meses de la cirugía inicial. De un 11-66% mueren de su enfermedad en los siguientes 5 años 62,156. - Metástasis a distancia (33%), a los 18 meses. Casi el 50% de los pacientes seguidos durante 24 meses desarrollan enfermedad sistémica y el 65-75% de los mismos morirán de su enfermedad140. Factores pronósticosSe consideran factores de importancia pronóstica 18,89,140,156 el estadio, el sexo y la edad del paciente, el tamaño y la localización del tumor, las características histológicas y la co-morbilidad por otras enfermedades o tumores no cutáneos. Las neoplasias cutáneas asociadas no empeoran el pronóstico 157. - Estadio El factor pronóstico más importante, íntimamente relacionado con la supervivencia libre de enfermedad y con la supervivencia global, es el estadio inicial de la enfermedad en el momento del diagnóstico, que pone de relieve la importancia de su diagnóstico precoz 8,12,21,100,158. En la serie de Allen et al 141 el estadio de la enfermedad en el momento del diagnóstico fue el único factor predictivo de supervivencia de los valorados en relación con el paciente, el tumor o el tratamiento. La presencia de afectación ganglionar es el factor predictivo más importante de supervivencia y de enfermedad metastásica a distancia. En una serie publicada, la media de supervivencia para pacientes con y sin afectación ganglionar fue de 13 versus 40 meses, respectivamente 159. En la serie de Allen et al 141, la supervivencia a los 5 años de pacientes con ganglios negativos fue de un 97% frente a un 52% de supervivencia en aquellos pacientes que debutaban con ganglios positivos. Por ello, estos mismos autores recomiendan la estadificación ganglionar mediante el estudio del ganglio centinela en los casos de CCM localizados al diagnóstico. Tal y como se ha señalado previamente, en su serie encontraron afectación ganglionar tras estudio anatomopatológico en un 25% de los pacientes con ganglios clínicamente negativos. La supervivencia a los 5 años de los pacientes con ganglios clínicamente negativos era del 75%, y la de los pacientes estadiados tras estudio del ganglio centinela del 97%. Además, la supervivencia a los 5 años disminuye con el número de ganglios afectos: de un 66% con un solo ganglio afecto a un 30% cunado existen más de 4 ganglios positivos. - Sexo y edad del paciente El sexo masculino se señala, en general, como de peor pronóstico que el femenino, aunque en algunas series no ha podido confirmarse 17. La edad al diagnóstico es un tema controvertido, aunque la mayor parte de las publicaciones coinciden en señalar un empeoramiento del pronóstico por encima de los 55-60 años. - Tamaño tumoral y localización La mayor parte de los autores están de acuerdo en que los tumores de > 2 cm tienen un peor pronóstico, aunque otros lo encontraron a partir de 3 cm 17 o de 5 cm 160 y Ott et al no hallaron significación estadística en el tamaño tumoral en su serie 14. La localización de la lesión primaria también influye en el pronóstico: las lesiones en el tronco, especialmente en la vulva y región perianal, tienen el peor pronóstico 14,53,66; las lesiones en las piernas tienen mayor tendencia a la recidiva, por la menor vascularización en pacientes ancianos, que impide una cirugía amplia, y la menor tolerancia a altas dosis de radioterapia 50. También se habló de un peor pronóstico en los tumores localizados en cabeza y cuello, quizás a causa de unos márgenes menos amplios por razones cosméticas 145. Los tumores en superficies mucosas también parecen tener un peor pronóstico por su mayor accesibilidad a los canales vasculares y linfáticos. - Características histológicas Los datos histológicos asociados tradicionalmente a un peor pronóstico son los tumores con células de pequeño tamaño y patrón difuso, un índice mitósico > 10 / campo de gran aumento, y la evidencia de invasión vascular y linfática. Los dos primeros factores son controvertidos, ya que la mayor parte de los tumores exhiben mezcla de los distintos subtipos histológicos, no habiéndose encontrado en algunas series significación pronóstica 17,161. Incluso, en una reciente publicación, Fernández-Figueras et al162 ha comunicado una inesperada asociación entre células de tamaño intermedio, alto índice de proliferación y recurrencias locales y/o metástasis. En cuanto al índice mitósico, algunos autores 17,162 recomiendan el empleo de un parámetro más preciso: la detección de Ki67, que expresada por más del 50% de las células implicaría un pronóstico desfavorable 17. La invasión linfovascular es un hecho habitual en los CCM y se observa con mayor frecuencia en los estadios más avanzados. El infiltrado inflamatorio se asocia para algunos autores con un pronóstico desfavorable, adquiriendo una elevada capacidad predictiva (86%), con una sensibilidad del 100% y una especificidad del 63% cuando se asocia a la profundidad de invasión del tumor (desfavorable cuando infiltra el tejido celular subcutáneo) 160. Para otros autores 17 el infiltrado inflamatorio es indicativo de una respuesta inmune y, por tanto, ligado a una evolución más favorable, como sucede en otros tumores, como el melanoma. Se ha sugerido por algunos autores que la expresión de CD44 99, y un estroma rico en laminina, colágeno IV e integrinas pueden econtrarse en relación con el riesgo de metástasis163, mientras que la expresión de bcl-2, p53 17,164 y c-myc 17,165 no se relacionan con este hecho. La expresión de CD117 en los CCM indica una mutación del c-kit, pero no ha podido establecerse relación con la progresión tumoral 17,166. SEGUIMIENTO | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
1 - Toker C. Trabecular carcinoma of the skin. Arch Dermatol 1972; 105:107-110. 2 - Tang CK, Toker C. Trabecular carcinoma of the skin: an ultrastructural study. Cancer 1978; 42: 2311-2321. 3 - Hoefler H, Kerl H, Rauch HJ, Denk H. New immunocytochemical observations with diagnostic significance in cutaneous neuroendocrine carcinoma. Am J Dermatopathol 1984; 6: 525-530. 4 - Wick MR, Scheithauer BW. En: Wick MR. Pathology of unusual malignant cutaneous tumors. New York: Marcel Dekker, 1985: 107-180. 5 - Krasagakis K, Almond-Roesler B, Zouboulis CC, Tebbe B, Wartenberg E, Wolff KD, Orfanos CE. Merkel cell carcinoma: report of ten cases with emphasis on clinical course, treatment, and in vitro drug sensitivity. J Am Acad Dermatol 1997; 36: 727-732. 6 - Boyle F, Pendlebury S, Bell D. Further insight into the natural history and management of primary cutaneous neuroendocrine (Merkel cell) carcinoma. Int J Radiat Oncol Biol Phys 1995; 31: 315-323. 7 - Chuang TY, Su WP, Muller SA. Incidence of cutaneous T cell lymphoma and other rare skin cancers in a defined population. J Am Acad Dermatol 1990; 23: 254-256. 8 - Medina-Franco H, Urist MM, Fiveash J, Heslin MJ, Bland KI, Beenken SW. Multimodality treatment of Merkel cell carcinoma: case series and literature review of 1024 cases. Ann Surg Oncol 2001; 8: 204-208. 9 - Hodgson NC. Merkel cell carcinoma: Changing incidence trends. J Surg Oncol 2005; 89: 1-4. 10 - Miller RW, Rabkin CS. Merkel cell carcinoma and melanoma: etiological similarities and diferences. Cancer Epidemiol Biomarkers Prev 1999; 8: 153-158. 11 - Penn I, First MR. Merkel's cell carcinoma in organ recipients: report of 41 cases. Transplantation 1999; 68: 1717-1721. 12 - Akhtar S, Oza KK, Wright J. Merkel cell carcinoma: report of 10 cases and review of the literature. J Am Acad Dermatol. 2000; 43: 755-767. 13 - Meyer-Pannwitt U, Kummerferldt K, Boubaris P, et al. Merkel cell tumor or neuroendocrine skin carcinoma. Langerbeks Arch Chir 1997; 382: 349-358. 14 - Ott MJ, Tanabe KK, Gadd MA, et al. Multimodality management of Merkel cell carcinoma. Arch Surg 1999; 134: 388-393. 15 - Koljonen V, Bohling T, Granhroth G, Tukiainen E. Merkel cell carcinoma: a clinicopathological study of 34 patients. Eur J Surg Oncol 2003; 29: 607-610. 16 - Paradela S, Peña C, Fernández-Jorge B, Vieira V, Rodríguez-Lozano J, García-Rozado A, Fonseca E. Carcinoma de células de Merkel. Actas Dermosifiliogr 2004; 95: 553-559. 17 - Llombart B, Monteagudo C, López-Guerrero JA, Carda C, Jorda E, Sanmartín O, Almenar S, Molina I, Martín JM, Llombart-Bosch A. Clinicopathological and immunohistochemical analysis of 20 cases of Merkel cell carcinoma in search of prognostic markers. Histopathology. 2005; 46: 622-634. 18 - Brenner B, Sulkes A, Rakowsky E, Feinmesser M, Yukelson A, Bar-Haim E, Katz A, Idelevich E, Neuman A, Barhana M, Fenig E. Second neoplasms in patients with Merkel cell carcinoma. Cancer. 2001; 91: 1358-1362. 19 - Pitale M, Sessions RB, Husain S. An analysis of prognostic factors in cutaneous neuroendocrine carcinoma. Laryngoscope 1992; 102: 244-249. 20 - Ratner D, Nelson BR, Brown MD, Johnson TM. Merkel cell carcinoma. J Am Acad Dermatol 1993; 29: 143-56. 21 - Kokoska, ER, Kokoska, MS, Collins, BT, Stapleton DR, Wade TP. Early aggressive treatment for Merkel cell carcinoma improves outcome. Am J Surg 1997; 174: 688-693. 22 - Walsh NM. Primary neuroencocrine (Merkel cell) carcinoma of the skin: morphologic diversity and implications thereof. Hum Pathol 2001; 32: 680-689. 23 - Lunder EJ, Stern RS. Merkel-cell carcinomas in patients treated with methoxsalen and ultraviolet A radiation. N Engl J Med 1998; 339: 1247-1248. 24 - Van Gele M, Kaghad M, Leonard JH, Van Roy N, Naeyaert JM, Geerts ML, Van Belle S, Cocquyt V, Bridge J, Sciot R, De Wolf-Peeters C, De Paepe A, Caput D, Speleman F. Mutation analysis of P73 and TP53 in Merkel cell carcinoma. Br J Cancer 2000; 82: 823-826. 25 - Kayashima K, Ono T, Johno M, Kojo Y, Yamashita N, Matsunaga W. Spontaneous regression in Merkel cell (neuroendocrine) carcinoma of the skin. Arch Dermatol 1991; 127: 550-553. 26 - Djilali-Bouzina F, Cribier B, Heid E. Regressive neuroendocirne carcinoma after partial biopsy. Nouv Dermatol 1992; 11: 767-770. 27 - Duncan WC, Ttschen JA. Spontaneous regression of Merkel cell (neuroendocrine) carcinoma of the skin. J Am Acad Dermatol 1993; 29: 653–654. 28 - Connelly TJ, Cribier B, Brown TJ, Yanguas I. Complete spontaneous regression of Merkel cell carcinoma: A review of the 10 reported cases. Dermatol Surg 2000; 26: 853-856. 29 - Maruo K, Kayashima KI, Ono T. Regressing Merkel cell carcinoma-a case showing replacement of tumour cells by foamy cells. Br J Dermatol 2000; 142: 1184-1189. 30 - Junquera L, Torre A, Vicente JC, García-Consuegra L, Fresno MF. Complete spontaneous regression of Merkel cell carcinoma. Ann Otol Rhinol Laryngol 2005; 114: 376-380. 31 - O'Rourque MG, Bell JR. Merkel cell tumor with spontaneous regression. J Dermatol Surg Oncol 1986; 12: 994-996. 32 - Bayrou O, Avril MF, Charpentier P, Caillou B, Guillaume JC, Prade M. Primary neuroendocrine carcinoma of the skin: clinico-pathologic study of 18 cases. J Am Acad Dermatol 1991; 24: 198-207. 33 - Connelly TJ, Kowalcyk AP. Another case of spontaneous regression of Merkel cell (neuroendocrine) carcinoma. Dermatol Surg. 1997; 23: 588-590. 34 - Yanguas I, Goday JJ, Gonzalez-Guemes M, Oleaga JM, Lozano M, Soloeta R. Spontaneous regression of Merkel cell carcinoma of the skin. Br J Dermatol 1997; 137: 296-298. 35 - Brown TJ, Jackson BA, Macfarlane DF, Goldberg LH. Merkel cell carcinoma: spontaneous resolution and management of metastatic disease. Dermatol Surg 1999; 25: 23-25. 36 - Gooptu C, Woollons A, Ross J, Price M, Wojnarowska F, Morris PJ, Wall S, Bunker CB. Merkel cell carcinoma arising after therapeutic immunosuppression. Br J Dermatol 1997; 137: 637-641. 37 - Lentz SR, Krewson L, Zutter MM. Recurrent neuroendocrine (Merkel cell) carcinoma of the skin presenting as marrow failure in a man with systemic lupus erythematosus. Med Pediatr Oncol 1993; 21: 137-141. 38 - Suzuki S, Koide M, Sakamoto S, Matsuo T. Pharmacokinetics of carboplatin and etoposide in a haemodialysis patient with Merkel-cell carcinoma. Nephrol Dial Transplant. 1997; 12: 137-40. 39 - Alonso A, Dauden E, Álvarez-Ruiz S, Ríos L, Fraga J, García-Díez A. Placa eritematosa frontal de crecimiento rápido. Actas Dermosifiliogr 2005; 96: 264-266. 40 - Rodríguez-Costa J, Rodríguez-Paternina E, Muñoz-Aguilera R. Metástasis de un carcinoma de células de Merkel en un paciente con transplante cardíaco, diagnosticado mediante PAAF. Patología 1996; 29: 255-258. 41 - Urbatsch A, Sams WM Jr, Urist MM, Sturdivant R. Merkel cell carcinoma occurring in renal transplant patients. J Am Acad Dermatol 1999; 41:289-291. 42 - Buell JF, Trofe J, Hanaway MJ, Beebe TM, Gross TG, Alloway RR, First MR, Woodle ES. Immunosuppression and Merkel cell cancer. Transplant Proc 2002; 34: 1780–81. 43 - An KP, Ratner D. Merkel cell carcinoma in the setting of HIV infection. J Am Acad Dermatol 2001; 45: 309-312. 44 - Engels EA, Frisch M, Goedert JJ, Biggar RJ, Miller RW. Merkel cell carcinoma and HIV infection. Lancet 2002; 359: 497-498. 45 - Silva EG, Mackay B, Goepfert H, Burgess MA, Fields RS. Endocrine carcinoma of the skin (Merkel cell carcinoma). Pathol Annu 1984; 19: 1-30. 46 - Valdés F, Sánchez-Aguilar D, Peteiro C, Toribio J. Carcinoma de células de Merkel y linfoma no Hodgkin. Actas Dermatosifiliogr 2002; 93: 247-249. 47 - Lien HC, Tsai TF, Lee YY, Hsiao CH. Merkel cell carcinoma and chronic arsenicism. J Am Acad Dermatol 1999; 41: 641-643. 48 - Haag ML, Glass LF, Fenske NA. Merkel cell carcinoma. Diagnosis and treatment. Dermatol Surg 1995; 21: 669-683. 49 - Ruiz R, Blasco J, Merino J, Linares J, Naranjo R. Carcinoma de células de Merkel. Presentación de seis casos. Actas Dermosifiliogr 2003; 94: 300-304. 50 - Poulsen M. Merkel-cell carcinoma of the skin. Lancet Oncol 2004; 5: 593-599. 51 - Coit DG. Merkel cell carcinoma. Ann Surg Oncol 2001;8: 99-102. 52 - Hapcic K, Panchal J, Stewart J, Levine N. Giant Merkel cell carcinoma involving the upper extremity. Dermatol Surg 2001; 27: 493-494. 53 - Tai PT, Yu E, Winquist E, Hammond A, Stitt L, Tonita J, Gilchrist J. Chemotherapy in neuroendocrine/Merkel cell carcinoma of the skin: case series and review of 204 cases. J Clin Oncol 2000; 18: 2493-2499. 54 - Woodworth B, Lacey JP, Amedee RG. Merkel cell carcinoma: an overview and case report. J La State Med Soc 2001; 153: 522-526. 55 - Brissett AE, Olsen KD, Kasperbauer JL, Lewis JE, Groellner JR, Spotts BE, et al. Merkel cell carcinoma of the head and neck: a retrospective series. Head Neck 2002; 24: 982-988. 56 - Chiarelli TG, Grant-Kels JM, Sporn JR, Rezuke WN, Whalen JD. Unusual presentation of a Merkel cell carcinoma. J Am Acad Dermatol 2000; 42: 366-370. 57 - Balaton AJ, Capron F, Baviera EE, Meyrignac P, Vaury P, Vuong PN. Neuroendocrine carcinoma (Merkel cell tumor?) presenting as a subcutaneous tumor. An ultrastructural and immunohistochemical study of three cases. Pathol Res Pract 1989; 184: 211-216. 58 - Goldenhersh MA, Prus D, Ron N, Rosenmann E. Merkel cell tumor masquerading as granulation tissue on a teenager's toe. Am J Dermatopathol 1992; 14: 560-563. 59 - Asioli S, Dorji T, Lorenzini P, Eusebi V. Primary neuroendocrine (Merkel cell) carcinoma of the nipple. Virchows Arch 2002; 440: 443-444. 60 - Tyring SK, Lee PC, Omura EF, Green Lk, Merot Y. Recurrent and metastatic cutaneous neuroendocrine (Merkel cell) carcinoma mimicking angiosarcoma. Arch Dermatol. 1987; 123: 1368-1370. 61 - Hitchcock, CL, Bland, KI, Laney RG, 3d, et al. Neuroendocrine (Merkel cell) carcinoma of the skin. Its natural history, diagnosis, and treatment. Ann Surg 1988; 207: 201-207. 62 - Shaw, JH, Rumball, E. Merkel cell tumour: clinical behaviour and treatment. Br J Surg 1991; 78: 138-142. 63 - Voog E, Biron P, Martin JP, Blay JY. Chemotherapy for patients with locally advanced or metastatic Merkel cell carcinoma. Cancer 1999; 85:2589-2595. 64 - Eusebi V, Capella C, Cossu A, Rosai J: Neuroendocrine carcinoma within lymph nodes in the absence of a primary tumor, with special reference to Merkel cell carcinoma. Am J Surg Pathol 1992, 16: 658-666. 65 - Díaz Iglesias JM, Fresno Forcelledo MF, Herrero-Zapatero A, Losa García JL, Díaz Iglesias C, Ablanedo Ablanedo P, Floriano Alvarez PL: Carcinoma de células de Merkel en ganglios linfáticos sin tumor cutáneo primario conocido. Presentación de 3 casos. Cirugía Española 1995; 57: 499-503. 66 - Poulsen M, Rischin D, Walpole E, Harvey J, Mackintosh J, Ainslie J, Hamilton C, Keller J, Tripcony L, Trans-Tasman Radiation Oncology Group. High-risk Merkel cell carcinoma of the skin treated with synchronous carboplatin/etoposide and radiation: a Trans-Tasman Radiation Oncology Group Study--TROG 96:07. J Clin Oncol 2003; 21: 4371-4376. 67 - Drijkoningen M, de Wolf-Peeters C, van Limbergen E, Desmet V. Merkel cell tumor of the skin: an immunohistochemical study. Hum Pathol. 1986; 17: 301-307. 68 - Leong AS, Phillips GE, Pieterse AS, Milios J. Criteria for the diagnosis of primary endocrine carcinoma of the skin (Merkel cell carcinoma). A histological, immunohistochemical and ultrastructural study of 13 cases. Pathology 1986; 18: 393-399. 69 - Gollard R, Weber R, Kosty MP, Greenway HT, Massullo V, Humberson C. Merkel cell carcinoma: review of 22 cases with surgical, pathologic, and therapeutic considerations. Cancer 2000; 88:1842-1851. 70 - Smith KJ, Skelton HG 3rd, Holland TT, Morgan AM, Lupton GP. Neuroendocrine (Merkel cell) carcinoma with an intraepidermal component. Am J Dermatopathol 1993; 15: 528-533. 71 - Ferringer T, Rogers HC, Metcalf JS. Merkel cell carcinoma in situ. J Cutan Pathol 2005; 32: 162–165. 72 - Sidhu GS, Feiner H, Flotte TJ, Mullins JD, Schaefler K, Schultenover SJ. Merkel cell neoplasms. Histology, electron microscopy, biology, and histogenesis. Am J Dermatopathol 1980; 2: 101-119. 73 - Frigerio B, Capella C, Eusebi V, Tenti P, Azzopardi JG. Merkel cell carcinoma of the skin: the structure and origin of normal Merkel cells. Histopathology 1983; 7: 229-249. 74 - Warner TF, Uno H, Hafez GR, Burgess J, Bolles C, Lloyd RV, Oka M. Merkel cells and Merkel cell tumors. Ultrastructure, immunocytochemistry and review of the literature. Cancer 1983; 52: 238-245. 75 - Fernández-Figueras MT, Puig L, Gilaberte M, Gómez-Plaza MC, Rex J, Ferrándiz C, Ariza A. Merkel cell (primary neuroendocrine) carcinoma of the skin with nodal metastasis showing rhabdomyosarcomatous differentiation. J Cutan Pathol 2002; 29: 619–622. 76 - Kossard S, Wittal R, Killingsworth M. Merkel cell carcinoma with a desmoplastic portion. Am J Dermatopathol 1995; 17: 517-522. 77 - Sibley RK, Dehner LP, Rosai J. Primary neuroendocrine (Merkel cell?) carcinoma of the skin. I. A clinicopathologic and ultrastructural study of 43 cases. Am J Surg Pathol. 1985; 9: 95-108. 78 - Goessling W, McKee PH, Mayer RJ. Merkel cell carcinoma. J Clin Oncol 2002; 20: 588-598. 79 - Freedberg RM, Eisen AZ, Wolff K, et al. Fitzpatrick’s Dermatology in General Medicine (ed 5). New York, NY, McGraw-Hill, 1990, pp 915-918, 1261, 2991. 80 - Takenaka H, Kishimoto S, Shibagaki R, Nagata M, Yasuno H. Merkel cell carcinoma with partial spontaneous regression: an immunohistochemical, ultrastructural, and TUNEL labeling study. Am J Dermatopathol 1997; 19: 614-618. 81 - Gould VE, Moll R, Moll I, Lee I, Franke WW. Neuroendocrine (Merkel) cells of the skin: hyperplasias, dysplasias, and neoplasms. Lab Invest 1985; 52: 334-353. 82 - Smith DF, Messina JL, Perrott R, Berman CG, Reintgen DS, Cruse CW, Glass FL, Fenske NA, DeConti RC, Trotti A 3rd. Clinical approach to neuroendocrine carcinoma of the skin (Merkel cell carcinoma). Cancer Control 2000; 7: 72-83. 83 - Khan Durani B, Hartschuh W. Merkel cell carcinoma. Clinical and histological diferential diagnosis, diagnostic approach and therapy. Hautarzt 2003; 54: 1171-1176. 84 - Miettinen M. Keratin 20: Immunohistochemical marker for gastrointestinal, urothelial, and Merkel cell carcinomas. Mod Pathol 1995; 8: 384-388. 85 - Chan JK, Suster S, Wenig BM, Tsang WY, Chan JB, Lau AL. Cytokeratin 20 immunoreactivity distinguishes Merkel cell (primary cutaneous neuroendocrine) carcinomas and salivary gland small cell carcinomas from small cell carcinomas of various sites. Am J Surg Pathol 1997; 21: 226-234. 86 - Heenan P J, Cole J M, Spagnolo D V. Primary cutaneous neuroendocrine carcinoma (Merkel cell tumor) an adnexal epithelial neoplasm. Am J Dermatopathol 1990: 12: 7–16. 87 - Mount SL, Taatjes DJ. Neuroendocrine carcinoma of the skin (Merkel cell carcinoma). An immunoelectron-microscopic case sutdy. Am J Dermatopathol 1994; 16: 60-65. 88 - Su LD, Lowe L, Bradford CR, et al. Immunostaining for cytokeratin 20 improves detection of micrometastatic Merkel cell carcinoma in sentinel lymph nodes. J Am Acad Dermatol 2002; 46: 661–666. 89 - Bickle K, Glass F, Messina JL, Fenske NA, Siegrist K. Merkel cell carcinoma: a clinical, histopathologic and immunohistochemical review. Semin Cutan Med Surg 2004; 23: 46-53 90 - Byrd-Gloster AL, Khoor A, Glass LF et al. Differential expression of thyroid transcription factor 1 in small cell lung carcinoma and Merkel cell tumor. Hum. Pathol. 2000; 31; 58–62 91 - Cheuk W, Kwan MY, Suster S, et al: Immunostaining for thyroid transcription factor 1 and Cytokeratin 20 aids the distinction of small cell carcinoma from Merkel cell carcinoma, but not pulmonary from extrapulmonary small cell carcinomas. Arch Pathol Lab Med 2001; 125: 228-231. 92 - Leech SN, Kolar AJ, Barrett PD, Sinclair SA, Leonard N. Merkel cell carcinoma can be distinguished from metastatic small cell carcinoma using antibodies to cytokeratin 20 and thyroid transcription factor 1. J Clin Pathol 2001; 54: 727-729. 93 - Moll I, Gillardon F, Waltering S, et al: Differences of bcl-2 protein expression between Merkel cells and Merkel cell carcinomas. J Cutan Pathol 1996; 23: 109-117. 94 - Sibley RK, Dahl D. Primary neuroendocrine (Merkel cell?) carcinoma of the skin. II. An immunocytochemical study of 21 cases. Am J Surg Pathol. 1985; 9: 109-116. 95 - Weiler R, Fischer-Colbrie R, Schmid KW, Feichtinger H, Bussolati G, Grimelius L, Krisch K, Kerl H, O'Connor D, Winkler H. Immunological studies on the occurrence and properties of chromogranin A and B and secretogranin II in endocrine tumors. Am J Surg Pathol 1988; 12: 877-884. 96 - Brinkschmidt C, Stolze P, Fahremkamp AG, et al. Immunohistochemical demonstration of chromogranin A, chromogranin B and secretoneurin in Merkel cell carcinoma of the skin: An immunohistochemical study suggesting two types of Merkel cell carcinoma. Appl Immunohistochem. 1995; 3: 37-44. 97 - Nicholson SA, McDermott MB, Swanson PE, Wick MR. CD99 and cytokeratin-20 in small-cell and basaloid tumors of the skin. Appl Immunohistochem Mol Morphol. 2000; 8: 37-41. 98 - Su LD, Fullen DR, Lowe L, Uherova P, Schnitzer B, Valdez R. CD117 (KIT receptor) expression in Merkel cell carcinoma. Am J Dermatopathol 2002; 24: 289-293. 99 - Penneys NS, Shapiro S. CD44 expression in the Merkel cell carcinoma may correlate with risk of metastasis. J Cutan Pathol 1994; 21: 22–26 100 - Smith PD, Patterson JW. Merkel cell carcinoma (neuroendocrine carcinoma of the skin). Am J Clin Pathol 2001; 115 Suppl: S68-78. 101 - Wilder RB, Harari PM, Graham AR, Shimm DS, Cassady JR. Merkel cell carcinoma. Improved locoregional control with postoperative radiation therapy. Cancer 1991; 68: 1004-1008. 102 - Szpak CA, Bossen EH, Linder J, Johnston WW. Cytomorphology of primary small-cell (Merkel-cell) carcinoma of the skin in fine needle aspirates. Acta Cytol 1984; 28: 290-296. 103 - Pettinato G, De Chiara A, Insabato L, Angrisani P, Saurel J, Morard JL, Ruocco V, Quarto F. Neuroendocrine (Merkel cell) tumor of the skin: fine-needle aspiration cytology, histology, electron microscopy and immunohistochemistry of 12 cases. Appl Pathol 1988; 6:17-27. 104 - Gattuso P, Castelli MJ, Shah PA, Kron T. Fine needle aspiration cytologic diagnosis of metastatic Merkel cell carcinoma in the parotid gland. Acta Cytol 1988; 32: 576-578. 105 - Gherardi G, Marveggio C, Stiglich F. Parotid metastasis of Merkel cell carcinoma in a young patient with ectodermal dysplasia. Diagnosis by fine needle aspiration cytology and immunocytochemistry. Acta Cytol 1990; 34: 831-836. 106 - al-Kaisi NK. Fine-needle aspiration cytology of a metastatic Merkel-cell carcinoma. Diagn Cytopathol 1991; 7: 184-188. 107 - Layfield LJ, Glasgow BJ. Aspiration biopsy cytology of primary cutaneous tumors. Acta Cytol 1993; 37: 679-688. 108 - Daskalopoulou D, Maounis N, Kokalis G, Liodandonaki P, Belezini E, Markidou S. The role of fine needle aspiration cytology in the diagnosis of primary skin tumors. Arch Anat Cytol Pathol 1993; 41: 75-81. 109 - Pérez-Guillermo M, Sola-Perez J, Abad-Montano C, Pastor Quirante FA, Montalban Romero MS. Merkel cell tumor of the eyelid and the cytologic aspect in fine-needle aspirates: report of a case. Diagn Cytopathol 1994; 10: 146-151. 110 - Gottschalk-Sabag S, Ne'eman Z, Glick T. Merkel cell carcinoma diagnosed by fine-needle aspiration. Am J Dermatopathol 1996; 18: 269-272. 111 - Collins BT, Elmberger PG, Tani EM, Bjornhagen V, Ramos RR. Fine-needle aspiration of Merkel cell carcinoma of the skin with cytomorphology and immunocytochemical correlation. Diagn Cytopathol 1998; 18: 251-257. 112 - Shin HJ, Caraway NP. Fine-needle aspiration biopsy of metastatic small cell carcinoma from extrapulmonary sites. Diagn Cytopathol 1998; 19: 177-181. 113 - Hallman JR, Shaw JA, Geisinger KR, Loggie BW, White WL. Cytomorphologic features of Merkel cell carcinoma in fine needle aspiration biopsies. A study of two atypical cases. Acta Cytol. 2000; 44: 185-93. 114 - Solomon RK, Lundeen SJ, Hamlar DD, Pambuccian SE. Fine-needle aspiration diagnosis of unusual cutaneous neoplasms of the scalp in HIV-infected patients: a report of two cases and review of the literature. Diagn Cytopathol 2001; 24: 186-192. 115 - Kabukcuoglu F, Sungun A, Polat N, Evren I, Kabukcuoglu Y. Fine needle aspiration cytology of Merkel cell carcinoma. Acta Cytol 2003; 47: 311-313. 116 - Dey P, Jogai S, Amir T, Temim L. Fine-needle aspiration cytology of Merkel cell carcinoma. Diagn Cytopathol 2004; 31: 364- 365. 117 - Domagala W, Lubinski J, Lasota J, Giryn I, Weber K, Osborn M. Neuroendocrine (Merkel cell) carcinoma of the skin. Cytology, intermediate filament typing and ultrastructure of tumor cells in fine needle aspiration. Acta Cytol 1987; 31: 267-75. 118 - Skoog L, Schmitt FC, Tani E. Neuroendocrine (Merkel-cell) carcinoma of the skin: immunocytochemical and cytomorphologic analysis on fine-needle aspirates. Diagn Cytopathol. 1990; 6: 53-57. 119 - Löwhajen T, Tani EM, Skoog L. Salivary glands and rare head and neck lesions. En: Bibbo M, editor. Comprehensive cytopathology. Chicago: Saunders, 1991; 127-133. 120 - Vortmeyer AO, Merino MJ, Boni R, Liotta LA, Cavazzana A, Zhuang Z. Genetic changes associated with primary Merkel cell carcinoma. Am J Clin Pathol 1998; 109: 565-570. 121 - Van Gele M, Leonard JH, Van Roy N, Van Limbergen H, Van Belle S, Cocquyt V, Salwen H, De Paepe A, Speleman F. Combined karyotyping, CGH and M-FISH analysis allows detailed characterization of unidentified chromosomal rearrangements in Merkel cell carcinoma. Int J Cancer. 2002; 101: 137-145. 122 - Leonard JH, Cook AL, Nancarrow D, Hayward N, Van Gele M, Van Roy N, Speleman F. Deletion mapping on the short arm of chromosome 1 in Merkel cell carcinoma. Cancer Detect Prev 2000; 24: 620-627. 123 - Leonard JH, Williams G, Walters MK, Nancarrow DJ, Rabitts PH. Deletion mapping of the short arm of chromosome 3 in Merkel cell carcinoma. Genes Chromosomes Cancer 1996; 15: 102-107 124 - Harnett PR, Kearsley JH, Dracopoli N. Loss of allelic heterozygosity on distal chromosome 1p in merkel cell carcinoma: a marker of neural crest origins. Cancer Genet Cytogenet 1991; 54: 109-113. 125 - Van Gele M, Speleman F, Vandesompele J, Van Roy N, Leonard JH. Characteristic pattern of chromosomal gains and losses in Merkel cell carcinoma detected by comparative genomic hybridization. Cancer Res 1998; 58: 503-1508. 126 - Plettenberg A, Pammer J, Tschachler E. Merkel cells and Merkel cell carcinoma express the BCL-2 proto-oncogene. Exp Dermatol 1996; 5: 183-188. 127 - Van Gele M, Leonard JH, Van Roy N, Cook AL, De Paepe A, Speleman F. Frequent allelic loss at 10q23 but low incidence of PTEN mutations in Merkel cell carcinoma. Int J Cancer. 2001; 92: 409-413. 128 - Stoppler H, Stoppler MC, Kisiela M, Holzbach A, Moll I, Houdek P, Moll R. Telomerase activity of Merkel cell carcinomas and Merkel cell carcinoma-derived cell cultures. Arch Dermatol Res 2001; 293: 397-406. 129 - Deichmann M, Kurzen H, Egner U, Altevogt P, Hartschuh W. Adhesion molecules CD171 (L1CAM) and CD24 are expressed by primary neuroendocrine carcinomas of the skin (Merkel cell carcinomas). J Cutan Pathol 2003; 30: 363-368. 130 - Kurokawa M, Nabeshima K, Akiyama Y, Maeda S, Nishida T, Nakayama F, Amano M, Ogata K, Setoyama M. CD56: a useful marker for diagnosing Merkel cell carcinoma. J Dermatol Sci 2003; 31: 219-224. 131 - Merkel F. Tastzellen and Tastkoerperchen bei den Hausthieren und beim Menschen. Arch Mikrosc Anat 1875; 11: 636-652. 132 - Krasagakis K, Tosca AD. Overview of Merkel cell carcinoma and recent advances in research. Int J Dermatol 2003, 42: 669–676 133 - Boot PM, Rowden G, Walsh N. The distribution of Merkel cells in human fetal and adult skin. Am J Dermatopathol. 1992; 14: 391–396. 134 - Halata Z, Grim M, Bauman KI. Friedrich Sigmund Merkel and his "Merkel cell", morphology, development, and physiology: review and new results. Anat Rec A Discov Mol Cell Evol Biol 2003; 271: 225-239. 135 - Gu J, Polak JM, Tapia FJ, Marangos PJ, Pearse AG. Neuron-specific enolase in the Merkel cells of mammalian skin. The use of specific antibody as a simple and reliable histologic marker. Am J Pathol 1981; 104: 63-68. 136 - Wick MR, Goellner JR, Scheithauer BW, Thomas JR 3rd, Sanchez NP, Schroeter AL. Primary neuroendocrine carcinomas of the skin (Merkel cell tumors). A clinical, histologic, and ultrastructural study of thirteen cases. Am J Clin Pathol 1983; 79: 6-13. 137 - Skelton HG, Smith KJ, Hitchcock CL, McCarthy WF, Lupton GP, Graham JH. Merkel cell carcinoma: analysis of clinical, histologic, and immunohistologic features of 132 cases with relation to survival. J Am Acad Dermatol 1997; 37: 734-739. 138 - Layfield L, Ulich T, Liao S, Barr R, Cheng L, Lewin KL. Neuroendocrine carcinoma of the skin: an immunohistochemical study of tumor markers and neuroendocrine products. J Cutan Pathol 1986; 13: 268-273. 139 - Gould E, Albores-Saavedra J, Dubner B, Smith W, Payne CM. Eccrine and squamous differentiation in Merkel cell carcinoma. An immunohistochemical study. Am J Surg Pathol 1988; 12: 768-772. 140 - Tai PT, Yu E, Tonita J, Gilchrist J. Merkel cell carcinoma of the skin. J Cutan Med Surg. 2000; 4: 186-195. 141 - Allen PJ, Bowne WB, Jaques DP, Brennan MF, Busam K, Coit DG. Merkel Cell Carcinoma: Prognosis and treatment of patients from a single institution. J Clin Oncol 2005; 23: 2300-2309. 142 - Shack RB, Barton RM, DeLozier J, Rees RS, Lynch JB. Is aggressive surgical management justified in the treatment of Merkel cell carcinoma?. Plast Reconstr Surg 1994; 94: 970-975. 143 - Marks ME, Kim RY, Salter MM. Radiotherapy as an adjunct in the management of Merkel cell carcinoma. Cancer 1990; 65: 60-64. 144 - Hohaus K, Kostler E, Schonlebe J, Klemm E, Wollina U. Merkel cell carcinoma: a retrospective analysis of 17 cases. JEADV 2003; 17: 20-24. 145 - O'Connor, WJ, Roenigk, RK, Brodland, DG. Merkel cell carcinoma. Comparison of Mohs micrographic surgery and wide excision in eighty-six patients. Dermatol Surg 1997; 23: 929-923. 146 - Boyer JD, Zitelli JA, Brodland DG, D'Angelo G. Local control of primary Merkel cell carcinoma: Review of 45 cases treated with Mohs micrographic surgery with and without adjuvant radiation. J Am Acad Dermatol 2002; 47: 885-892. 147 - Victor S, Morton B, Smith J. Merkel cell carcinoma: is prophylactic lymph node dissection indicated? Am Surg 1996; 62: 879–882. 148 - Pan D, Narayan D, Ariyan S. Merkel cell carcinoma: five case reports using sentinel lymph node biopsy and a review of 110 new cases. Plast Reconstr Surg 2002; 110: 1259–1265. 149 - Zeitouni, NC, Cheney, RT, Delacure, MD. Lymphoscintigraphy, sentinel lymph node biopsy, and Mohs micrographic surgery in the treatment of Merkel cell carcinoma. Dermatol Surg 2000; 26: 12-18. 150 - Rodrigues LK, Leong SP, Kashani-Sabet M, Wong JH. Early experience with sentinel lymph node mapping for Merkel cell carcinoma. J Am Acad Dermatol 2001; 45: 303-308. 151 - Westgate SJ. Radiation therapy for skin tumors. Otolaryngol Clin North Am 1993; 26: 295-309. 152 - Muller A, Keus R, Neumann N, Lammering G, Schnabel T. Management of Merkel cell carcinoma: case series of 36 patients. Oncol Rep 2003; 10: 577-585. 153 - Carpentier O, Carrotte-Lefebvre I, Patenotre P, Mirabel X, Delaporte E, Piette F. Primitive cutaneous neuroendocrine carcinomas or Merkel tumor. Clinical and therapeutic aspects in 22 patients. Presse Med 2002; 31: 735-739. 154 - Brown HA, Sawyer DM, Woo T. Intraepidermal Merkel cell carcinoma with no dermal involvement. Am J Dermatopathol. 2000; 22: 65-69. 155 - Fenig E, Brenner B, Katz A, Rakovsky E, Hana MB, Sulkes A. The role of radiation therapy and chemotherapy in the treatment of Merkel cell carcinoma. Cancer 1997; 80: 881–885. 156 - Eng TY, Boersma MG, Fuller CD, Cavanaugh SX, Valenzuela F, Herman TS. Treatment of merkel cell carcinoma. Am J Clin Oncol 2004; 27: 510-515. 157 - Bahadir S, Çobanoglu Ü, Alpay K, Özoran Y, Parlat P, Aykanat D. Merkel cell carcinoma. J Eur Acad Dermatol Venerol. 2003; 17: 602-603. 158 - Benessahraoui M, Dalstein V, Lorchel F, Algros MP. Puzenat E. Louvat P. Hassam B. Humbert PH. Aubin F. Carcinoma à cellules de Merkel: etude descriptive de 24 cas. Rev Med Interne 2003; 24: 560-568. 159 - Morrison WH, Peters LJ, Silva EG, Wendt CD, Ang KK, Goepfert H. The essential role of radiation therapy in securing locoregional control of Merkel cell carcinoma. Int J Radiat Oncol Biol Phys 1990; 19: 583-591. 160 - Mott RT, Smoller BR, Morgan MB. Merkel cell carcinoma: a clinicopathologic study with prognostic implications. J Cutan Pathol. 2004; 31; 217–223. 161 - Pilotti S, Rilke F, Bartoli C, Grisotti A. Clinicopathologic correlations of cutaneous neuroendocrine Merkel cell carcinoma. J Clin Oncol 1988; 6: 1863–1873. 162 - Fernández-Figueras MT, Puig L, Musulen E, Gilaberte M, Ferrándiz C, Lerma E, Ariza A. Prognostic significance of p27Kip1, p45Skp2 and Ki67 expression profiles in Merkel cell carcinoma, extracutaneous small cell carcinoma, and cutaneous squamous cell carcinoma. Histopathology. 2005; 46, 614–621. 163 - Sames K, Schumacher U, Halata Z, et al. Lectin and proteoglycan histochemistry of Merkel cell carcinomas. Exp Dermatol. 2001; 10:100-106. 164 - Kennedy MM, Blessing K, King G, Kerr KM. Expression of bcl-2 and p53 in Merkel cell carcinoma. An immunohistochemical study. Am J Dermatopathol 1996; 18: 273–277. 165 - Jemec C, Ghana J. Merkel cell carcinoma: survival and oncogens markers. J Eur Acad Dermatol Venerol. 2000; 14: 400-404. 166 - Feinmesser M, Halpern M, Kaganovsky E, Brenner B, Fenig E, Hodak E, Sulkes J, Okon E. c-kit expression in primary and metastatic Merkel cell carcinoma. Am J Dermatopathol. 2004; 26: 458-462. 167 - Bertó J, Cuenca A, Díaz-Martínez B, Peña ML, Ruíz-Fernández P, Sánchez de Paz F. Carcinoma de células de Merkel. Estudio de cinco casos. Actas Dermosifiliogr 2005; 96: 106-110. 168 - Yiengpruksawan A, Coit DG, Thaler HT, Urmacher C, Knapper WK. Merkel cell carcinoma. Prognosis and management. Arch Surg 1991; 126: 1514-1519. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Rabu, 15 Desember 2010
Carcinoma de células de Merkel




Langganan:
Posting Komentar (Atom)
Tidak ada komentar:
Posting Komentar